

Abstract
Understanding how proteins are organized in compartments is essential to elucidating their function. While proximity-dependent approaches such as BioID have enabled a massive increase in information about organelles, protein complexes, and other structures in cell culture, to date there have been only a few studies on living vertebrates. Here, we adapted proximity labeling for protein discovery in vivo in the vertebrate model organism, zebrafish. Using lamin A (LMNA) as bait and green fluorescent protein (GFP) as a negative control, we developed, optimized, and benchmarked in vivo TurboID and miniTurbo labeling in early zebrafish embryos. We developed both an mRNA injection protocol and a transgenic system in which transgene expression is controlled by a heat shock promoter. In both cases, biotin is provided directly in the egg water, and we demonstrate that 12 h of labeling are sufficient for biotinylation of prey proteins, which should permit time-resolved analysis of development. After statistical scoring, we found that the proximal partners of LMNA detected in each system were enriched for nuclear envelope and nuclear membrane proteins and included many orthologs of human proteins identified as proximity partners of lamin A in mammalian cell culture. The tools and protocols developed here will allow zebrafish researchers to complement genetic tools with powerful proteomics approaches.
Keywords: Proximity-dependent biotinylation, TurboID, miniTurbo, zebrafish embryo, BioID
Abbreviations: AMP, adenosine monophosphate; APEX, enhanced ascorbate peroxidase; BASU, Bacillus subtilis biotin ligase; BFDR, Bayesian false discovery rate; BioID, proximity-dependent biotin identification; FDR, false discovery rate; GFP, green fluorescent protein; GO, gene ontology; HPF, hours post fertilization; LMNA, lamin A; MS, mass spectrometry; WT, wild type
Graphical Abstract
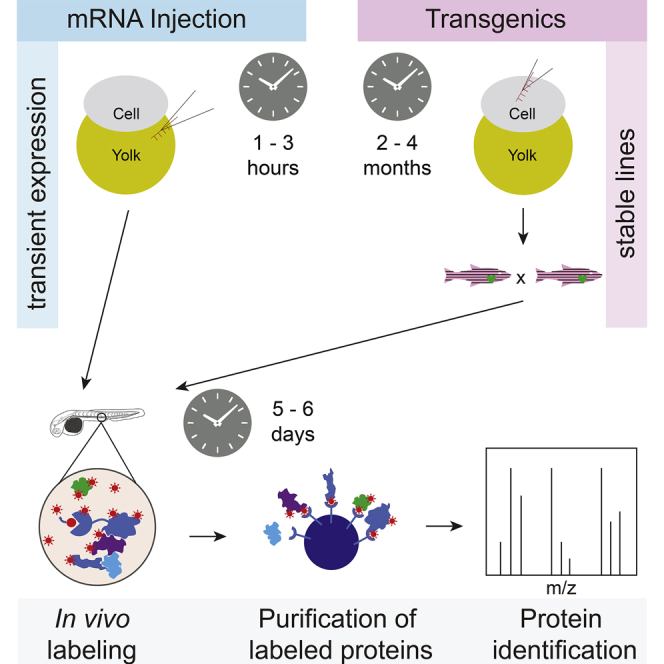
Highlights
-
•
Implementation of a proximity-dependent biotinylation in zebrafish embryos.
-
•
Use of miniTurbo and TurboID for proximal interaction detection in zebrafish embryos.
-
•
Optimization of mRNA injection and transgenics-based enzyme delivery.
-
•
Benchmarking conditions and approaches on the nuclear lamina.
In Brief
Proximity-dependent biotinylation approaches such as APEX and BioID are used to illuminate cellular organization in cell culture studies but are not often applied to whole organisms. Here, Rosenthal et al. report the development and optimization of a toolkit for proximity-dependent biotinylation with miniTurbo and TurboID in zebrafish embryos. They describe, using the lamin A component of the lamina, protocols and benchmarks for a rapid injection-based strategy that is appropriate for early development and a flexible heat shock inducible transgenic system.
Probing the organization of proteins into complexes, organelles, and other structures in the context of living organisms is critical to understanding the context-dependent nature of protein function. While unicellular model organisms, particularly the yeast Saccharomyces cerevisiae, have been well characterized, the organization of the proteome in the living cells of multicellular organisms including vertebrates is still largely uncharted. The vertebrate model organism Danio rerio (zebrafish) is a popular model to interrogate vertebrate development. While traditionally used for developmental studies due to its high fecundity, external fertilization, rapid development, and optical clarity during early development (1), recently the use of zebrafish for disease modeling and drug screening has increased. This has spurred a coincident increase in genetic tools adapted or developed for zebrafish use. However, the adaptation and development of tools for proteomic studies in zebrafish have lagged behind.
In zebrafish, there are two main methods by which exogenous protein expression can be driven, either by the injection of mRNA or through the generation of transgenic lines (reviewed in (2)). Each approach has its strengths and limitations. Injection of in vitro transcribed mRNA is usually performed at the single cell stage to achieve expression throughout the developing embryo. The advantage is that there is no lag time in performing an experiment as there would be when generating a transgenic animal. This method is most appropriate when early development (<72 h post fertilization (hpf)) is being studied, as consistent expression levels are difficult to maintain for much longer due to mRNA and protein degradation (3). A potential drawback, particularly in cases where many embryos are required, is that each embryo needs to be injected individually, a time-consuming task and one potentially leading to variability of protein expression levels. Finally, tissue-specific expression is not possible via mRNA injection. Conversely, transgenics are ideal for later time points and should afford versatility in both expression levels and site of expression when employing different promoters, e.g., stage-specific or tissue-specific (reviewed in (4)). Disadvantages of transgenics are the relatively lengthy generation time of 2.5 to 4 months and the resultant overall difficulty in scaling up the number of genes profiled by the approach.
Studies of proteome organization have typically employed biochemical fractionation and affinity purification, both coupled to mass spectrometry. These methods are ideal for studying stable interactions after tissues and cells have been lysed, but they require that the protein complexes or organelles stay intact throughout the purification (5). Additionally, the cellular context is disrupted upon lysis, which leads to loss of information as well as the risk for postlysis artifacts.
The introduction of proximity-dependent biotinylation techniques in 2012 has offered new possibilities for the detection of protein–protein interactions, including weaker interactions, and the definition of the composition of both membrane-bound and membraneless organelles (reviewed in (6)). Proximity-dependent biotinylation approaches include biotin ligase-based methods such BioID (7) and peroxidase-based methods such as APEX (8) that permit the covalent labeling of preys proximal to a protein of interest in the context of living cells. Biotin ligase-based methods take advantage of a mutated biotin ligase, either an E. coli BirA harboring a single point mutation [R118 G, referred to as BirA∗ in the original BioID study (7)], molecularly evolved derivatives called miniTurbo and TurboID (9)—see below—or mutated biotin ligases from other species such as BioID2 from Aquifex aeolicus (10) or BASU from Bacillus subtilis (11). These mutated enzymes are capable of activating biotin to the reactive intermediate biotinyl-5′-AMP, but instead of completing the transfer of biotin to a biotin-acceptor peptide, they release it from the active site where it can react with primary amines in the vicinity, resulting in their covalent biotinylation (reviewed in (12)). Fusion of the mutated biotin ligase to a protein of interest (the “bait”), expression of the fusion in a cell of interest, and addition of exogenous biotin therefore lead to the labeling of proteins within a ~10 nm radius from the bait (13). The labeled proteins (preys) include a mixture of direct and indirect interactors (often including weak or cycling interacting partners) as well as proteins that reside in the same locale as the bait (collectively referred to as “proximal interactors”). Because the biotin is covalently bound, harsh lysis, solubilization, and washes can be performed, allowing for the recovery of previously inaccessible proximal interactors.
Proximity-dependent biotinylation has been employed by numerous groups to investigate the proximal interactomes of proteins, complexes, and organelles in cells grown in culture. While BioID with the original BirA∗ enzyme (7) or the A. aeolicus biotin ligase BioID2 (10) has been performed in unicellular and multicellular organisms such as Dictyostelium discoideum (14), Plasmodium berghei (15), Toxoplasma gondii (16), Trypanosoma brucei (17), Saccharomyces cerevisiae (18), and Nicotiana benthamiana (19), there have been relatively few reports of their use in vertebrates, most of them in mice (20, 21, 22, 23, 24, 25). Pronobis et al. (26) used BioID2 to study heart regeneration in transgenic zebrafish following repeated injections of biotin intraperitoneally over 3 days, demonstrating that zebrafish is an amenable model for proximity-dependent biotinylation.
Major hurdles to the application of BioID with first-generation enzymes in larger animal models relate both to the relatively poor activity of these enzymes (forcing long labeling times as in the zebrafish BioID2 experiment (26)) and the need to deliver supplemental biotin to the appropriate tissues or organs. The generation of molecularly evolved variants of the E. coli BirA enzyme, called TurboID and miniTurbo, that display 15 to 30 times the labeling efficiency of BirA∗ has been a major breakthrough in enabling more efficient proximity-dependent biotinylation in different in vivo models, including C. elegans (9), D. melanogaster (9, 27), A. thaliana (28), N. benthamiana (28, 29), S. pombe (30), and M. musculus (31). Using transgenics in zebrafish, Xiong et al. (32) recently demonstrated the applicability of TurboID through the fusion of a GFP nanobody to TurboID: crosses to transgenic lines expressing fusions of proteins of interest to the Clover green fluorescent protein (GFP) enabled labeling and recovery of proximal interactors to cavin proteins. Yet, in vivo proximity-dependent biotinylation approaches are still in their infancy, and more thorough optimization and benchmarking of the approaches are needed; it has also not been clearly determined whether the more rapid and scalable mRNA injection systems can lead to high-quality proximal proteomes.
Here, we have adapted proximity-dependent biotinylation with the miniTurbo and TurboID enzymes for use in live zebrafish embryos and demonstrate its versatility as a method that can be applied both via mRNA injection and generation of transgenic animals. To benchmark our method, we demonstrate that we can identify known proximal interactors of the nuclear envelope protein, lamin A. This establishes a toolbox and methodology where any bait can be examined in a rapid fashion in zebrafish embryos via the mRNA injection-based approach, or more slowly but with greater nuance using the transgenic approach. This will be of particular value for proteins whose function is cell- or context-dependent.
Experimental Procedures
Generation of Zebrafish Vector Sequences and Expression Constructs
The TurboID and miniTurbo sequences (9) were optimized for zebrafish expression using the online codon optimization tool from Integrated DNA Technologies (IDT). A zebrafish consensus Kozak sequence shown to generate high expression (33) was added at the start site (GCAAACatgGCG). A flexible linker (GS2) was included downstream of the protein coding sequence followed by a 3× FLAG tag. Finally, a Gateway cloning attR1 site was added at the 3′ end of the sequences. Upstream and downstream restriction sites were also included to allow for restriction cloning.
The above constructs were synthesized and cloned into the pUC57 vector (General Biosystems Inc). A fusion construct was built in the pcDNA5-pDEST vector. Briefly, the synthesized constructs were restriction cloned (NheI/NotI) into the pcDNA5-pDEST vector upstream of Gateway cloning attR2 sites. A Gateway-compatible entry clone for LMNA (Genbank accession number EU832167, encoding human lamin A) was transferred into the pcDNA5-pDEST Gateway destination vector using LR Gateway cloning. This resulted in a mammalian expression vector of LMNA tagged with TurboID or miniTurbo at its N-terminus. Vectors were constructed in the identical fashion for controls using EGFP (enhanced green fluorescent protein), by subcloning the EGFP cassette from pDONR233-eGFP (itself a derivative of pEGFP-C1, BD Biosciences). All the constructs were validated by restriction digest and DNA sequencing of the subcloned fragments.
The cassettes (TurboID or miniTurbo fused to LMNA or GFP) were amplified by PCR from pcDNA5-pDEST and transferred to vectors compatible with zebrafish expression. First, restriction cloning was used to transfer the cassettes into the pCS2+ vector for mRNA expression (SP6-based) for injection in zebrafish. Briefly, PCR amplified cassettes were digested with ClaI and XbaI and ligated into the pCS2+ vector downstream of an SP6 promoter sequence and upstream of an SV40 polyA cassette. The vectors were validated by restriction digest and DNA sequencing of the subcloned fragments.
Alternatively, the generation of constructs for transgenic generation used the Gateway compatible Tol2kit (34). The Tol2kit is a transposon-based transgenesis method, whereby Gateway cloning is used to insert fragments containing 3′, middle, and 5′ inserts into a destination vector containing flanking Tol2 transposon recognition sites. Upon injection of the vector together with mRNA coding for the Tol2 transposase, the cassette is randomly inserted into the genome (34). The cassettes (TurboID or miniTurbo fused to LMNA or GFP) were amplified by PCR as above and inserted into the Gateway compatible Tol2kit middle entry vector pME-MCS (34) via restriction cloning as above. This vector contains the multiple cloning site derived from the pBluescript II SK+ vector inserted into the pDONR221 vector (34). Tol2 destination vectors were generated using three-fragment Gateway cloning. The Tol2kit destination vector pDestTol2CG2 (34) was used for all constructs as it includes a cardiomyocyte-specific GFP expression sequence, allowing for visual selection of integration positive embryos. A three-fragment Gateway LR reaction was used to generate the final destination vectors. At the 5′ end, the Tol2kit p5E-HSP70l (34) vector was used to donate the 1.5 kb HSP70l promoter fragment (which enables heat shock-inducible expression). This was followed by the fusion protein sequence cassette (TurboID or miniTurbo fused to LMNA or GFP) obtained from the pME-MCS vector. Lastly, an SV40 polyA sequence was obtained from the Tol2kit P3E-polyA (34) vector. All the constructs were validated by restriction digest and DNA sequencing of the subcloned fragments.
Zebrafish Husbandry
Zebrafish were maintained under the guidance and approval of the Canadian Council on Animal Care and the Hospital for Sick Children Laboratory Animal Services. Embryos were maintained at 28.5 °C in embryo medium (egg water). When used, biotin supplementation was performed by directly dissolving biotin powder (BioBasic, cat. BB0078) in heated (~90 °C) egg water followed by cooling and adjustment of the pH to 7. Wild type (WT) or transgenic fish of the strain TL/AB were used for all experiments.
mRNA Injection-Based Expression of Fusion Proteins
For expression of fusion proteins via mRNA injection, the pCS2+ vectors containing the fusion constructs were linearized by digestion with NotI. Capped mRNA was generated from the linearized vector using the mMESSAGE mMACHINE SP6 Transcription Kit (ThermoFisher Scientific cat. AM1340) and the transcribed mRNA purified using the MEGAclear Transcription Clean-Up Kit (ThermoFisher Scientific cat. AM1908). The mRNA was quantified by spectrophotometry (NanoDrop 2000) and run on an agarose gel to determine that only a single band was present (to ensure degradation had not occurred). For protein expression, 400 pg of capped mRNA was injected into the yolk of single cell stage WT embryos. For western blots, a pool of ~100 deyolked (see below “Preparation of embryos for protein extraction”) embryos at 48 h post fertilization (hpf) was used to generate lysate, with 50 μg of pooled lysate (equivalent to ~10 embryos) run on the gel per sample. For TurboID pull-downs, ~1000 and ~200 deyolked embryos were used for 12 and 48 hpf collections, respectively. In order to properly synchronize the staging of the pool of ~1000 12 hpf embryos, the injections and collections were staggered. Approximately 250 embryos were injected per mating tank over ~15 min, after which the injections were repeated with a new mating tank. The injected embryos were incubated at 28.5 °C in biotin-supplemented egg water and collected 12 h after their respective injection times. For TurboID, all experiments were performed in biological replicates (duplicates or triplicates as described in the relevant figures), with separate injections performed on different days.
Generation of Transgenic Zebrafish Lines and Protein Expression
To generate transgenic zebrafish lines, 30 pg of the final Tol2 destination vectors (purified using the QIAprep Spin Miniprep Kit; Qiagen cat. 27106) was injected into single cell stage zebrafish embryos together with 75 pg of Tol2 transposase mRNA (purified using the MEGAclear Transcription Clean-Up Kit; Thermo Fisher Scientific cat. AM1908). The injected embryos were screened for positive construct integration based on the presence of GFP expression in the heart at 24 to 48 hpf (4). GFP-positive embryos were grown to adulthood. To determine if the GFP positive adults had germ-line integration of the constructs, the fish were outcrossed to WT fish and the progeny screened for GFP expression in the heart. These F1 GFP-positive progeny were grown to adulthood. Embryos from incrosses of F1 adults were used for experiments.
Embryos were kept at 28.5 °C until induction of transgene expression by heat shock. At the selected time point for transgene induction (60 hpf), the embryos were transferred to 50 ml conical tubes filled with egg water preheated to 38 °C and placed in a 38 °C water bath for 1 h (35). Following heat shock, the embryos were returned to plates with 28.5 °C egg water and incubated for the times indicated. Previous studies have found mRNA expression to be induced as early as 15 min post heat shock, while protein expression follows at 1.5 to 4 h post heat shock. While the stability of the mRNA transcript varies greatly (degrading as soon as 3 h post heat shock), protein expression is generally more stable (36, 37, 38).
Preparation of Embryos for Protein Extraction
Following protein expression via mRNA injection or heat shock induction, labeling was allowed to proceed at 28.5 °C. Labeling was stopped by washing the embryos in ice-cold egg water. Embryos were dechorionated using pronase in egg water (1 mg/ml; Roche, Cat# 10165921001). Embryos were washed three times with ice-cold egg water and transferred to 1.5 ml microcentrifuge tubes. Embryos were deyolked essentially as described (39). Briefly, embryos were washed twice with ice-cold deyolking buffer (55 mM NaCl, 1.8 mM KCl, 1.25 mM NaHCO3). In total, 200 μl deyolking buffer supplemented with 1 mM phenylmethylsulfonyl fluoride (PMSF; BioShop Canada, Cat# PMS123) and protease inhibitor cocktail (Sigma-Aldrich, Cat# P8340, 1:100) were added, and the yolk was disrupted by gentle pipetting using a p200 tip. The yolk was further solubilized by vortexing three times for 2 s. The embryos were spun down (300g for 30 s) and the supernatant was removed. Embryos were washed twice with ice-cold wash buffer (110 mM NaCl, 3.5 mM KCl, 2.7 mM CaCl2, 10 mM Tris-HCl pH 8.5). Following removal of the supernatant, the embryos were snap frozen in liquid nitrogen and stored at −80 °C.
Immunoblotting
For western blot analysis of mRNA injected embryos, 50 μg of pooled lysate from ~100 48 hpf embryos was used, while for transgenic lines, 50 μg of pooled lysate from ~100 72 hpf embryos was used. Protein extraction was performed as for TurboID affinity purification (see below) with the following modification: the volume employed was 1 μl per embryo for 48 and 72 hpf embryos. A BCA assay (ThermoFisher Scientific, Cat# 23227) was used to quantify protein concentration in the lysate. The lysate was boiled in Laemmli SDS-PAGE sample buffer for 5 min and 50 μg of protein per lane was separated onto 10% SDS-PAGE gels and transferred onto nitrocellulose membranes (GE Healthcare Life Science, Cat# 10600001). Following Ponceau S staining, blots were blocked in 4.5% milk (for anti-BirA probing) or 5% bovine serum albumin (for Streptavidin-Horseradish peroxidase (HRP) probing) in TBS with 0.1% Tween-20 (TBST). Bait proteins were probed using rabbit anti-BirA antibody (Sino Biological, Cat# 11582-RP01) at 1:2000 in block buffer, washed in TBST, and detected with 1:5000 Donkey anti-Rabbit IgG-HRP. Biotinylated proteins were probed using Streptavidin-HRP (GE Healthcare Life Science, Cat# RPN1231) at 1:2000 in blocking buffer. Membranes were developed using ECL Western Blotting Detection Reagents (GE Healthcare Life Science, Cat# RPN2109). Quantification of western blots was performed using the Image Studio Lite software (Version 5.2, LI-COR Biosciences)
Live Imaging and Whole Mount Immunofluorescence
For imaging GFP expression post heat shock, embryos were subjected to heat shock at 38 °C for 1 h. At 6 h post heat shock, the embryos were sedated by the addition of tricaine methanesulfonate (0.01 mg/ml) (Sigma-Aldrich, Cat# A5040) and were imaged using a Zeiss Axio ZoomV16 Stereo microscope. Following imaging, the embryos were placed in fresh egg water.
Embryos that were to be used in immunofluorescence experiments were grown in egg water supplemented with 0.003% 1-phenyl 2-thiourea (PTU) (Sigma-Aldrich, Cat# P7629) from 24 hpf to prevent pigmentation. Following induction by heat shock of protein expression at 60 hpf and 12 h of labeling, 72 hpf embryos were collected for immunofluorescence. Fixing and imaging were performed essentially as in reference (40), with the following modifications. Embryos were washed three times in ice-cold egg water prior to fixation. Fixed embryos were washed twice in PBS +0.1% Tween-20 (PBSTw) prior to stepwise dehydration using methanol in PBSTw (25%, 50%, 75%, 100% methanol). Following rehydration (performed as dehydration but in reverse), permeabilization was performed with ice-cold acetone at −20 °C for 20 min, followed by three 10 min washes with PBS +1% Triton-X100. Embryos were blocked with 10% Normal Goat Serum (NGS; Millipore, Cat# S26-l) and 5% Bovine Serum Albumin (BSA) in PBS +0.4% Triton-X100 (PBSTx) for 1.5 h at room temperature before being stained for bait protein with mouse anti-FLAG antibody (Monoclonal anti-FLAG M2 antibody; Sigma-Aldrich, Cat# F3165; 1:500) in blocking buffer overnight at 4 °C. Embryos were washed six times for 30 min in PBSTx. For secondary detection, embryos were stained with goat anti-mouse Alexa Fluor 488 (Molecular Probes, Thermo Fisher Scientific, A-11032; 1:200) and for visualizing protein biotinylation, with Alexa Fluor 594 streptavidin conjugate (Molecular Probes, Thermo Fisher Scientific, S21374; 1:500). DAPI (1:1000) was used as a nuclear counterstain. All secondary staining was performed in blocking buffer overnight at 4 °C. Embryos were washed six times for 30 min in PBSTx and stored at 4 °C in PBSTw. For imaging, embryos were mounted in 1% low melt agarose and imaged with a Nikon A1R Si Point Scanning Confocal microscope.
TurboID Affinity Purification and Mass Spectrometry
For TurboID, the affinity purification followed essentially the protocol described in (41). Briefly, embryos were lysed in modified RIPA (modRIPA) buffer [50 mM Tris-HCl, pH 7.4, 150 mM NaCl, 1 mM EGTA, 0.5 mM EDTA, 1 mM MgCl2, 1% NP40, 0.1% SDS, 0.4% sodium deoxycholate, 1 mM PMSF, and 1× Protease Inhibitor mixture]. The volumes employed were 0.5 μl per embryo for 12 hpf embryos and 2 μl per embryo for 48 and 72 hpf embryos. Embryos were sonicated for 15 to 30 s at 30 to 35% amplitude (5 s on, 3 s off for three cycles at 30% amplitude for 12 hpf embryos, 10 s on, 3 s off for three cycles at 32% amplitude for 48 hpf embryos, and 10 s on, 3 s off for three cycles at 35% amplitude for 72 hpf embryos) on a Q500 Sonicator with 1/8” Microtip (QSonica, Newtown, Connecticut, Cat# 4422). Following sonication, 250 U of TurboNuclease (BioVision Inc, Cat# 9207) was added and the samples were incubated at 4 °C with rotation for 20 min. Next 10% SDS was added to the sample to bring the SDS concentration to 0.4%, and the sample was incubated at 4 °C with rotation for a further 5 min. Samples were centrifuged at 16,000g for 20 min and the supernatant was used for affinity purification of biotinylated proteins using 25 μl of washed streptavidin agarose beads (GE Healthcare Life Science, Cat# 17511301) for 16 h with rotation at 4 °C. The beads were next washed once with SDS-Wash buffer (25 mM Tris-HCl, pH 7.4, 2% SDS), twice with RIPA (50 mM Tris-HCl, pH 7.4, 150 mM NaCl, 1 mM EDTA, 1% NP40, 0.1% SDS, 0.4% sodium deoxycholate), once with TNNE buffer (25 mM Tris-HCl, pH 7.4, 150 mM NaCl, 1 mM EDTA, 0.1% NP40), and three times with 50 mM ammonium bicarbonate (ABC buffer), pH 8.0. Proteins were digested on-bead with 1 μg of trypsin (Sigma Aldrich, Cat# 6567) in 50 μl ABC buffer with rotation at 37 °C overnight. A further 0.5 μg of trypsin was added, and digestion continued for 3 h. Following digestion, the beads were spun down (400g for 30 s) and the supernatant was transferred to a new tube. The beads were washed twice with 75 μl water and the wash water was collected and added to the supernatant. A final spin was performed at 10,000 rpm for 2 min, and the supernatant (less than 15 μl to ensure no beads are transferred) was transferred to a new tube. The supernatant was acidified with 0.1 volume of 50% formic acid and dried by vacuum centrifugation. Peptides were resuspended in 5% formic acid and stored at −80 °C.
Mass Spectrometry Acquisition
Digested peptides were analyzed by nanoLC-MS/MS in a Data-Dependent Acquisition (DDA) workflow. Tryptic peptides from 200 embryos (at 48 and 72 HPF) or 900 embryos (at 12 HPF) were used, and 2/3 of each sample was injected for DDA analysis. Packed column emitters with a 5 to 8 μm tip opening were generated from 75 μm internal diameter fused silica capillary tubing, using a laser puller (Sutter Instrument Co, model P-2000). Nano-spray emitters were packed with C18 reversed-phase material (Reprosil-Pur 120 C18-AQ, 3 μm) resuspended in methanol using a pressure injection cell to a packed length of 15 cm. Samples were analyzed using an Eksigent 425 nanoHPLC connected to a Thermo Fusion Lumos Mass Spectrometer. Samples were loaded directly onto the packed tip emitter using the autosampler at a flow rate of 400 nl/min. Peptides were eluted from the column using a 90 min gradient (2–30% acetonitrile) at 200 nl/min. After 90 min, the acetonitrile concentration was increased to 80% over 10 min, held for 5 min, and then the column was equilibrated at 2% acetonitrile for 30 min. The DDA method used a 240k resolution and 5e5 target setting for MS1 scans. A 3 s cycle was used for HCD MS/MS acquired with 1 Da isolation, 15k resolution, 2e5 target, and 50 ms max fill time with a dynamic exclusion set to 12 s.
Mass Spectrometry Data Analysis
Mass spectrometry data generated were stored, searched, and analyzed using ProHits laboratory information management system (LIMS) platform (42). Within ProHits, ProteoWizard (V3.0.1072) (43) was used to convert the .RAW files to .mgf and .mzML formats. The data files were searched using Mascot (V2.3.02) (44) and Comet (V2016.01 rev.2) (45) against zebrafish sequences from the RefSeq database (version 65, July 02, 2014), supplemented with “common contaminants” from the Max Planck Institute (http://www.coxdocs.org/doku.php?id=maxquant:start_downloads.htm) and the Global Proteome Machine (GPM; ftp://ftp.thegpm.org/fasta/cRAP/crap.fasta), sequence tags (BirA, GST26, mCherry and GFP), LysC, and streptavidin. Reverse (decoy) entries were created for all sequences for a total of 86,384 entries. Search parameters were set to search for tryptic cleavages, allowing two missed cleavage sites per peptide a mass tolerance of 12 ppm and a tolerance of 0.15 amu for fragment ions. Variable modifications included deamidation (asparagine and glutamine) and oxidation (methionine). Results from each search engine were analyzed through TPP (the Trans-Proteomic Pipeline, v.4.7 POLAR VORTEX rev 1) (46) via the iProphet pipeline (47). All proteins with an iProphet probability ≥95% and two unique peptides were used for analysis.
Interaction Scoring
SAINTexpress (Version 3.6.1.) (48) was used to score the probability that identified proteins were enriched above background. SAINTexpress uses a model whereby each prey identified in a TurboID experiment with a given bait is compared using spectral counts as a measure of abundance, against a set of negative controls. For each separate experiment (different bait or condition), independent biological replicates were used to generate the prey profile, and this profile was compared against negative controls. A minimum of two biological replicates were used for all analysis. When three purifications were performed for a given bait and conditions, two virtual replicates were generated prior to SAINTexpress scoring. The virtual replicates were generated by selecting the two highest spectral counts for each prey in the purification of the bait across the three replicates. Creating these virtual replicates increases sensitivity when the reproducibility across the biological replicates is not perfect. Negative controls consisted of purifications from embryos expressing TurboID-GFP (six and three replicates for the TurboID mRNA injection and transgenic experiments, respectively) or miniTurbo-GFP (five and three replicates for the miniTurbo mRNA injection and transgenic experiments, respectively) and from embryos not expressing any exogenous fusion proteins (three replicates for all experiments). For running SAINTexpress, the 6 to 9 negative controls were compressed to four virtual controls, meaning that the four highest spectral counts for each identified protein were used in the scoring to increase stringency. Scores were averaged across both biological replicates (or virtual replicates), and these averages were used to calculate a Bayesian False Discovery Rate (BFDR); preys detected with a BFDR of ≤1% were considered high-confidence.
Human Orthology
To compare our zebrafish data with human data, we converted the zebrafish protein names to their human orthologs. We used the g:profiler g:Convert tool (49) and manual curation from NCBI (50) to make the conversions.
Gene Ontology Enrichment Analysis
Gene ontology (GO) enrichment analysis was performed using the human orthologs of the identified high-confidence proximal interactors as inputs. The g:Profiler g:GOSt tool (49) was used for the enrichment analysis. Default options were used, with the exceptions that the data source was set to only “GO cellular component terms,” with term size between 5 and 500.
Comparison With Previously Published Datasets
To compare our data with previously published datasets, we obtained the lists of high-confidence LMNA proximal interactors from the supplemental data of the Samavarchi-Tehrani et al. (51) and May et al. (52) publications. Using these lists of high-confidence proximal interactors, we performed GO enrichment analysis in the same manner as for our own data (see above). For comparison to the interactors listed on BioGRID, the full curated list of interactors for the H. sapiens LMNA protein was obtained from the BioGRID website (https://thebiogrid.org/110186/summary/homo-sapiens/lmna.html, accessed April 04, 2021) and compared against the high-confidence proximal interactors identified our experiments.
Data Visualization
Dot plots, scatter plots, and Pearson correlation data were generated using ProHits-viz (53). In the ProHits-viz dot plots tool, once a prey passes the selected FDR threshold (≤1% used here) with one bait, all its quantitative values across all baits are recovered. The FDR of the prey is then indicated by the edge color. Quantitative information (spectral count) is represented by the color gradient within the node, while relative prey counts across all baits are denoted by the size of the node (i.e., the maximal size for each prey corresponds to the bait in which it was detected with the highest number of spectra).
Experimental Design and Statistical Rationale
For each TurboID experiment, independent biological duplicates or triplicates were used. Each replicate was generated through independent mRNA injections or egg lays (for transgenic experiments) and collections. Statistical scoring using SAINTexpress against 6 to 9 controls compressed to four virtual controls was performed as described above (“Interaction Scoring”). The control samples used were TurboID-GFP or miniTurbo-GFP matched to the enzyme and expression method used in the experiment, and WT embryos not expressing exogenous protein, used in all experiments and age/treatment matched to the experiment unless otherwise noted. The same WT control runs were used in both TurboID and miniTurbo analysis. The Bayesian FDR was calculated using the average SAINTexpress score across replicates. The SAINTexpress analysis was performed independently for each enzyme and expression method. In other words, each of the following sets was analyzed independently: a) the mRNA injection TurboID-LMNA 12 and 48 h labeling runs, b) the mRNA injection miniTurbo-LMNA 12 and 48 h labeling runs, c) the transgenic TurboID-LMNA, d) the transgenic miniTurbo-LMNA, and e) the transgenic TurboID-LMNA comparison of labeling conditions.
Results
Adapting TurboID and miniTurbo for In vivo Biotinylation in Zebrafish
To determine whether the TurboID and miniTurbo enzymes could efficiently biotinylate proteins in early zebrafish embryos, codon-optimized gene constructs of TurboID and miniTurbo were synthesized for fusion to proteins of interest. The human lamin A (LMNA) bait was selected for optimization experiments as it has a well-characterized proximal interactome in mammalian cells (7, 51, 52) as well as a characteristic subcellular localization, enabling visual evaluation of the functionality of the fusions. This bait was first used to demonstrate the usefulness of the BioID method in identifying the proximal proteome of insoluble proteins that are typically difficult targets for conventional protein–protein interaction methods (7). The human and zebrafish LMNA proteins share 62% identity and 76% similarity at the amino acid level, and mutations of conserved sites result in similar protein mislocalization in both zebrafish and human cells (54, 55). This suggested that the human LMNA protein would function and localize appropriately when expressed in zebrafish. As a negative control, the GFP was subcloned in the same system.
To test the biotinylation efficiency of TurboID and miniTurbo in zebrafish, we used mRNA injections in zebrafish embryos to express TurboID or miniTurbo fused to LMNA or GFP coding sequences. Briefly, capped mRNA for TurboID and miniTurbo fusions were generated by in vitro transcription, purified, and injected into single cell stage zebrafish embryos (Fig. 1A). The injected embryos were grown in egg water supplemented with biotin for 12 or 48 h. Labeling was stopped by washing the embryos with ice-cold egg water without biotin. The embryos were dechorionated, deyolked, washed, and frozen for later analysis.
Fig. 1.

mRNA injection-based expression identifies LMNA proximal proteins in the early zebrafish embryo.A, in vitro transcribed mRNA coding for TurboID fused to GFP was injected into the single cell stage zebrafish embryo and GFP fluorescence was visualized at 6, 12, and 48 hpf (magnification 16×). B, TurboID-LMNA or miniTurbo-LMNA was expressed by mRNA injections (with the corresponding GFP fusions used as negative controls). Injected embryos were collected at 12 and 48 hpf. The number of proteins detected at BFDR ≤1% under each of the conditions is shown. C, list of the 18 high-confidence proximal interactors shared by all four conditions in (B). D, the 25 most abundant proteins (by spectral counts) with a BFDR ≤1% from the 12 h TurboID-LMNA condition are listed, alongside their abundance across each of the conditions.
To determine what level of biotin supplementation was required to achieve optimal protein biotinylation, we injected TurboID-LMNA and miniTurbo-LMNA mRNA into 100 embryos per condition and incubated them in egg water supplemented with increasing biotin concentrations (0, 200, and 800 μM) for 48 h. In total, 50 μg total lysate (equivalent to approximately ten embryos) per condition was separated onto SDS-PAGE gels and used to assess the extent of total biotinylation using streptavidin-horseradish peroxidase (Strep-HRP) blots. The levels of expressed tagged proteins were detected using an anti-BirA antibody capable of detecting TurboID and miniTurbo fusions. In the absence of any biotinylation enzyme injection (uninjected embryos), several biotinylated bands were readily detected in the absence of biotin, and biotin supplementation increased this signal ~1.9-fold at the highest concentration used. Injection of TurboID-LMNA (and to a lesser extent, miniTurbo-LMNA) increased the signal up to ~2.2-fold in the absence of biotin and up to ~2.7-fold after biotin addition. Many of the bands were in common to all conditions, indicating that they are background proteins (such background proteins have been well-documented in cell culture and notably include endogenously biotinylated proteins such as mitochondrial carboxylases (56, 57)). However, some of the bands appear specific to the TurboID and miniTurbo conditions (supplemental Fig. S1A). We note that the high signal detected with TurboID in the absence of biotin is consistent with reports that it has a high avidity for biotin and is capable of scavenging even small concentrations of this metabolite (9). We also note that the miniTurbo protein was detected at low levels, with an increase in detection upon the addition of biotin (supplemental Fig. S1B). These findings are consistent with reports of miniTurbo instability and biotin-dependent stabilization (52, 58). We did not notice any obvious developmental delay or morphological deformity with either concentration of biotin or injection of miniTurbo or TurboID fusions, suggesting low toxicity in our system (not shown). We therefore selected the highest (800 μM, near the upper limit of biotin solubility in water) concentration of biotin tested for the next set of experiments.
Characterization of the Lamin A Proximal Interactome Following mRNA Injection in Zebrafish
To explore whether the signal-to-noise was sufficient to identify proximity partners for LMNA in zebrafish embryos (or whether the high background was limiting the usefulness of the system), we next purified biotinylated proteins via streptavidin pull-down and identified them by mass spectrometry. To assess whether biotinylation was specific for our protein of interest, we compared the proximal proteome of LMNA (fused to either TurboID or miniTurbo) to that of GFP (fused with the same enzymes) and also purified biotinylated proteins from uninjected embryos (WT). Briefly, embryos (900 per replicate for the 12 h time point, and 200 for 48 h) were injected with the indicated mRNA. Following injection, embryos were incubated in biotin-supplemented egg water for labeling, for 12 or 48 h. The 12 h labeling time covers development through gastrulation to the start of somitogenesis (59), while the 48 h labeling time covers early development until the hatching stage at the completion of much of primary organogenesis (59). Labeling was stopped by washing embryos in ice-cold egg water. To enrich for biotinylated proteins, streptavidin pull-downs were performed on lysates from pools of mRNA-injected embryos, and nonbiotinylated peptides were eluted by tryptic digestion. Released peptides were identified by mass spectrometry, and peptides were assigned to proteins by the Trans-Proteomic Pipeline iProphet (iProphet) tool (46, 47). Proteins identified with an iProphet probability ≥95% and two unique peptides were used for analysis. Experiments were performed using biological replicates (duplicates for 12 h and triplicates for 48 h), with the following exceptions: for the negative control TurboID-GFP four replicates were used, for uninjected embryos only one replicate was used at 12 h and two at 48 h. Between 412 proteins (uninjected, 48 h) and 1279 proteins (TurboID-LMNA, 48 h) were identified per replicate (Table 1). Across all experiments, the 48 h labeling yielded a higher number of identified proteins than the 12 h labeling, as expected. TurboID yielded 23 to 29% more identifications than the corresponding miniTurbo constructs, with the exception of the 48 h GFP controls where identifications were almost equal (Table 1). For all labeling times, the proteins detected with the highest spectral counts were shared between the LMNA and GFP baits, suggesting that they are nonspecific (background) proteins. At the 12 h time point, these consisted mainly of mitochondrial and yolk proteins, while at 48 h yolk protein detection was reduced and muscle proteins became highly abundant.
Table 1.
Unique proteins identified by mRNA injection-based BioID experiments
| Experiment | Replicate | Parameter |
|||
|---|---|---|---|---|---|
| Unique proteins identified | Total Unique Proteins identified | Shared unique proteins identified | Proteins with a SAINT BFDR ≤1% | ||
| TurboID-LMNA 12 h Labeling |
Rep 1 | 774 | 982 | 616 | 89 |
| Rep 2 | 824 | ||||
| TurboID-LMNA 48 h Labeling |
Rep 1 | 1010 | 1708 | 668 | 144 |
| Rep 2 | 1279 | ||||
| Rep 3 | 1173 | ||||
| miniTurbo-LMNA 12 h Labeling |
Rep 1 | 759 | 854 | 557 | 37 |
| Rep 2 | 642 | ||||
| miniTurbo-LMNA 48 h Labeling |
Rep 1 | 970 | 1273 | 495 | 34 |
| Rep 2 | 727 | ||||
| Rep 3 | 851 | ||||
| TurboID-GFP 12 h Labeling |
Rep 1 | 864 | 1163 | 701 | N/A |
| Rep 2 | 1000 | ||||
| TurboID-GFP 48 h Labeling |
Rep 1 | 1278 | 1586 | 492 | N/A |
| Rep 2 | 714 | ||||
| Rep 3 | 867 | ||||
| Rep 4 | 1032 | ||||
| miniTurbo-GFP 12 h Labeling |
Rep 1 | 751 | 927 | 628 | N/A |
| Rep 2 | 804 | ||||
| miniTurbo-GFP 48 h Labeling |
Rep 1 | 1076 | 1525 | 565 | N/A |
| Rep 2 | 756 | ||||
| Rep 3 | 1199 | ||||
| Uninjected 12 h Labeling |
Rep 1 | 621 | N/A | N/A | N/A |
| Uninjected 48 h Labeling |
Rep 1 | 412 | 844 | 325 | N/A |
| Rep 2 | 757 | ||||
The number of unique proteins identified by ≥2 unique peptides and an iProphet probability ≥95% are listed for all mRNA injection-based experiments. For LMNA bait experiments, the number of proteins with a SAINT BFDR ≤1% are listed.
We next applied SAINTexpress (SAINT) (48) to identify proteins specific to the LMNA fusion conditions. SAINTexpress analysis of this dataset was performed as follows. First, for the 48 h labeling, we combined the triplicate purifications of the baits into two virtual replicates (i.e., the two highest spectral counts for each prey in the purification of the bait across the three replicates are selected); this increases sensitivity when the reproducibility across the biological replicates is not perfect (see Experimental Procedures). We also combined using the same approach the eight (for miniTurbo) or nine (for TurboID) negative controls into four virtual controls (as in (60)); this step increases the stringency in SAINTexpress scoring. The controls consisted of uninjected embryos (duplicate of 48 hpf embryos and one run of 12 hpf embryos) and six or five replicates of TurboID or miniTurbo, respectively (two replicates at 12 hpf for both enzymes, four and three replicates at 48 hpf for TurboID-GFP and miniTurbo-GFP respectively). All negative control samples were incubated with 800 μM biotin for the duration of the experiment. We are considering as high-confidence proximal partners those that pass a BFDR cutoff of ≤1%. This revealed 89 and 144 high-confidence proximal interactors identified at 12 and 48 h, respectively, for TurboID-LMNA and 37 and 34 proximal interactors with miniTurbo-LMNA (Table 1 and supplemental Table S1). Of these unique proteins, 40 were identified by both TurboID-LMNA and miniTurbo-LMNA (at either time points; Fig. 1B), with 17 identified across all conditions (Fig. 1C). The 20 proteins with the highest spectral counts and a BFDR ≤1% in the TurboID-LMNA 12 h labeling condition included proteins known to localize to the inner nuclear membrane, components of the nuclear pore complex, and previously identified LMNA proximal partners (Fig. 1D). From this list, all the proteins annotated to the inner nuclear membrane were readily detected across all conditions. The nuclear pore complex proteins displayed greater differences across conditions, with the 48 h labeling time producing a lower relative abundance with either TurboID or miniTurbo (Fig. 1D).
Correlation between replicates is an important metric of the reproducibility of the labeling experiments and is an essential component of the SAINT scoring (i.e., only preys enriched across replicates are considered significant). For post SAINT correlations, we used a BFDR ≤5% as a cutoff, to include proteins just missing the ≤1% BFDR cutoff. The 12 h labeling time resulted in very high correlation between replicates, with pairwise correlation pre and post SAINT ranging from 0.87 to 0.98 (supplemental Fig. S2). As expected (due to the increased complexity of the tissue and the longer labeling time allowing labeling of more background proteins), with 48 h of labeling there was greater variability, with pairwise correlation between replicates lower than with 12 h of labeling (range 0.36–0.89; supplemental Fig. S3). We conclude that for most applications, 12 h of labeling is sufficient and will allow for easier filtering, using fewer control samples. Forty-eight hour labeling produces higher background, but not significantly higher labeling of proximal interactors, thereby reducing the overall sensitivity after background contaminant filtering.
Taken together, these results indicate that, using mRNA injection as an expression method, TurboID-LMNA and miniTurbo-LMNA specifically biotinylate proximal proteins at a level high enough to allow for identification above background.
Generation of Inducible Transgenic Zebrafish Lines for In vivo TurboID
Having shown that proximity-dependent biotinylation and proximal protein identification could be achieved in zebrafish embryos, we progressed to performing these experiments in inducible stable transgenic zebrafish. This method has the advantage of allowing control of labeling via inducible expression of the transgene at any time point during development. To allow for temporal control of expression, we selected the hsp70l heat shock inducible promoter, which is well tolerated by zebrafish embryos (61), and does not require drug treatment. Using the Tol2 transgenesis system, stable transgenic zebrafish were generated to express, under the control of the 1.5 kb hsp70l promoter, TurboID, or miniTurbo fused to either LMNA or GFP as a control, resulting in the generation of four lines. All protein fusions included a 3× FLAG tag in addition to the biotin ligase to enable detection of the expressed protein and immunoprecipitation. We designate these transgenic lines: Tg(hsp70l:TurboID-LMNA)hsc158, Tg(hsp70l:TurboID-GFP)hsc159, Tg(hsp70l:miniTurbo-LMNA)hsc160, and Tg(hsp70l:miniTurbo-GFP)hsc161. All transgenic constructs also included GFP expressed under the control of the cardiomyocyte specific myl7 promoter (34), allowing for the identification and selection of integration-positive embryos by visual inspection of robust GFP fluorescence in the heart, generally assessed at 48 hpf, but visible as early as 16 hpf (Fig. 2A).
Fig. 2.

Inducible transgenic expression of TurboID-LMNA or miniTurbo-LMNA identifies LMNA proximal proteins.A, cassettes compatible with the Tol2 trangenesis system were generated that express the selected fusion proteins (TurboID-LMNA and GFP, miniTurbo-LMNA and GFP) downstream of the heat shock inducible Hsp70 promoter. The cassettes also contain GFP under the control of a cardiomyocyte specific Cmlc2 promoter to allow selection of integration-positive embryos. B, TurboID-GFP and miniTurbo-GFP transgenic embryos were heat shocked at 60 hpf for 1 h and imaged at 72 hpf. Integration-positive embryos were selected prior to heat shock based on expression of GFP in the heart (yellow arrow). GFP expression 12 h following heat shock is indicated by white arrowheads, and GFP expression in the eye by red arrows (magnification 16×, Scale Bar = 1 mm). C, immunofluorescence microscopy on embryos collected at 72 hpf following 1 h heat shock at 60 hpf time point was performed as detailed in Experimental Procedures. FLAG staining to detect the bait expression and streptavidin-fluorophore (Strep) to detect biotinylation. DAPI was used to stain the nucleus. (Upper panels Scale bar 50 μm, magnification 20×; Lower panels scale bar 5 μm, magnification 40×). D, the scatterplot shows all proteins detected with a SAINTexpress Bayesian FDR ≤1% with all least one of the baits (miniTurbo on the y-axis, TurboID on the x-axis); the dashed lines represent the different fold changes listed at the top. Preys are color-coded based on uniquely scoring as high-confidence interactors with either miniTurbo (green) or TurboID (blue) while preys scoring as high-confidence with both baits are coded in red. E, list of the 27 common partners. F, the 25 most abundant proteins (by spectral counts) with a BFDR ≤1% from the TurboID-LMNA experiment are listed, alongside their abundance across both conditions.
Expression was induced by heating zebrafish embryos to 38 °C for 1 h prior to returning the embryos to their normal incubation temperature (28.5 °C) for 11 h. The heat shock induced low levels of visible fluorescence by 3 h post-initiation, as visualized by TurboID-GFP fluorescence, with robust ubiquitous expression visible from 5 h through to collection of embryos at 12 h post heat shock. GFP expression is expected in the eye even without heat shock since the hsp70l promoter endogenously drives expression in the eye lens during early development (62) (Fig. 2B).
To ensure that the fusion proteins were localizing correctly and that protein biotinylation was occurring specifically proximal to the fusion protein, whole mount immunofluorescence was performed to determine the localization of TurboID-LMNA, miniTurbo-LMNA, TurboID-GFP, miniTurbo-GFP and to define where biotinylation occurred. An anti-FLAG antibody was used to visualize the fusion proteins and streptavidin conjugated to a fluorophore was employed to visualize biotinylation. Embryos were incubated in water supplemented with 800 μM biotin. Protein expression was induced by heat shock at 60 hpf for 1 h at 38 °C. Following heat shock, the embryos were incubated for 12 h before collection at 72 hpf. Immunofluorescence showed that TurboID-LMNA and miniTurbo-LMNA are localized, as expected, around the nucleus. Biotinylated proteins were also localized surrounding the nucleus, suggesting that biotinylation was occurring specifically proximal to the fusion proteins (note that this biotinylation signal may include the bait itself). This contrasted with TurboID-GFP and miniTurbo-GFP where GFP and biotinylation were visible throughout the cell. In all samples, background streptavidin staining was evident in the mitochondria (Fig. 2C).
Inducible Expression of TurboID and miniTurbo in Transgenic Zebrafish Embryos Can Be Used to Identify Proximal Partners
Having shown that the inducible fusion protein is properly localized (and leads to biotinylation in the expected locale), we moved to performing TurboID-based proteomics in transgenic zebrafish embryos. We wanted to use the inducible transgenic lines to perform TurboID experiments outside the time window possible with mRNA injections. For this reason, we elected to induce transgene expression at 60 hpf. We also selected the shortest labeling time we had tested with mRNA injection, 12 h, as this short labeling time would demonstrate the utility of this system for tracking developmental processes over time. This labeling period corresponds to the final stages of zebrafish embryonic growth, with the larval stage beginning at 72 hpf (59).
For all experiments, protein expression was induced by heat shock of the embryos at 38 °C for 1 h. Embryos expressing TurboID and miniTurbo fused to GFP and WT embryos were used as controls. The SAINTexpress software (SAINT) was used to filter the identified protein lists. By comparing against the control protein lists and between replicates, filtered lists of proteins unique to the experimental condition were generated.
For our initial transgenic TurboID experiments, TurboID-LMNA and miniTurbo-LMNA expression was induced at 60 hpf, labeling continued for 12 h, and the embryos were collected and labeling terminated at 72 hpf. The embryos were incubated in egg water supplemented with 800 μM biotin for the full 72 h. This prolonged biotin supplementation prior to induction of transgene expression was selected to ensure sufficient time for biotin accumulation within the embryo, as some polar compounds can require extended incubation to achieve high internal concentrations in zebrafish embryos (63). Following affinity purification and mass spectrometry, a total of 662 and 692 proteins were identified across triplicates for TurboID-LMNA and miniTurbo-LMNA, respectively (Table 2).
Table 2.
Unique proteins identified by heat shock inducible transgenic-based BioID experiments
| Experiment | Replicate | Parameter |
|||
|---|---|---|---|---|---|
| Unique proteins identified | Total unique proteins identified | Shared unique proteins identified | Proteins with a SAINT BFDR ≤1% | ||
| HSP:TurboID-LMNA 12 h Labeling |
Rep 1 | 1123 | 1453 | 662 | 64 |
| Rep 2 | 1191 | ||||
| Rep 3 | 824 | ||||
| HSP:miniTurbo-LMNA 12 h Labeling |
Rep 1 | 1133 | 1515 | 764 | 59 |
| Rep 2 | 1098 | ||||
| Rep 3 | 1105 | ||||
| HSP:TurboID-GFP 12 h Labeling |
Rep 1 | 1171 | 1600 | 697 | N/A |
| Rep 2 | 1336 | ||||
| Rep 3 | 853 | ||||
| HSP:miniTurbo-GFP 12 h Labeling |
Rep 1 | 716 | 1482 | 513 | N/A |
| Rep 2 | 1034 | ||||
| Rep 3 | 1157 | ||||
| WT 12 h Labeling |
Rep 1 | 1086 | N/A | N/A | N/A |
The number of unique proteins identified by ≥2 unique peptides and an iProphet probability ≥95% are listed for all heat shock inducible transgenic experiments. All experiments used 1 h heat shock (38 °C) induction at 60 hpf, and collection at 72 hpf. For LMNA bait experiments, the number of proteins with a SAINT BFDR ≤1% are listed.
SAINTexpress analysis of this dataset was performed as for the mRNA injection dataset, with the triplicates combined into two virtual replicates and the six controls combined into four virtual controls. The controls consisted of WT embryos (duplicate of 48 hpf embryos and one run of embryos heat shocked at 60 hpf and collected at 72 hpf) and triplicates of TurboID or miniTurbo fused to GFP (heat shocked at 60 hpf and collected at 72 hpf). All negative control samples were incubated with 800 μM biotin for the duration of the experiment. Correlation between replicates ranged from 0.27 to 0.80 prior to SAINTexpress scoring and from 0.36 to 0.81 when considering only hits with a BFDR ≤5% (supplemental Fig. S4; we note that these correlations are much lower than what we observe when analysis is performed with human cells in culture with any proximity-dependent biotinylation enzyme, where we routinely get correlations >0.9, e.g., (51, 64)). Following SAINTexpress scoring, 64 and 59 proteins with a BFDR ≤1% remained for TurboID-LMNA and miniTurbo-LMNA, respectively, with 27 of them shared (Fig. 2, D and E; supplemental Table S2). Seventeen of these 27 shared proteins were among the 25 most abundant (after SAINT filtering) high-confidence proximal interactors (by spectral count) identified by TurboID-LMNA with a BFDR ≤1% (Fig. 2, D and F).
Gene Ontology Enrichment Analysis
To determine if the identified high-confidence proximal interactors were enriched for proteins known to localize near LMNA, we performed GO enrichment analysis. We first converted the zebrafish genes to their human orthologs to take advantage of the greater degree of annotations (supplemental Table S3). GO profiling of the human orthologs of the identified proximal interactors with a BFDR ≤1% found “Nuclear Membrane” and “Nuclear Envelope” to be the most highly enriched Cellular Component (CC) categories (CC categories containing 5–500 terms). This was true for both TurboID and miniTurbo in both mRNA injection and transgenic experiments (supplemental Table S4). For mRNA injection-based experiments, between 18 and 32 of the high-confidence proximal interactors were annotated with these terms. For TurboID-LMNA and miniTurbo-LMNA inducible transgenic TurboID experiments, these terms annotated 26 and 18 high-confidence proximal interactors, respectively (Table 3, these numbers are higher than the direct output from g:Profiler since some annotated genes are duplicated in the zebrafish genome and both proteins were detected), with 14 of them detected with both enzymes. These results demonstrate that both mRNA injection-based and inducible transgenic expression of TurboID-LMNA and miniTurbo-LMNA can specifically biotinylate proximal proteins at a sufficiently high level as to achieve consistent identification above background (Fig. 3, A and B). A large proportion (24–55%) of the high-confidence proximal interactors in all experimental conditions are annotated as nuclear envelope or nuclear membrane proteins, supporting the conclusion that the labeling is occurring specifically proximal to the bait (Table 3).
Table 3.
Summary of protein identifications for all zebrafish BioID experiments and comparison to previously published datasets
| Result parameter | Condition (age) |
|||||
|---|---|---|---|---|---|---|
| TurboID-LMNA transgenic 12 h labeling (72 hpf) | miniTurbo-LMNA transgenic 12 h labeling (72 hpf) | TurboID-LMNA injected 12 h labeling (12 hpf) | TurboID-LMNA injected 48 h labeling (48 hpf) | miniTurbo-LMNA injected 12 h labeling (12 hpf) | miniTurbo-LMNA injected 48 h labeling (48 hpf) | |
| Proteins Identified (Total SAINT input) | 1452 | 1514 | 981 | 1707 | 853 | 1272 |
| Proteins Identified (SAINT FDR ≤1%) | 64 | 59 | 89 | 144 | 37 | 34 |
| Number of Human Orthologs | 60 | 55 | 85 | 133 | 35 | 33 |
| BioGRID Overlap LMNA Interactors | 24 (40%) | 20 (36.4%) | 41 (48.2%) | 45 (33.8%) | 19 (54.3%) | 15 (45.5%) |
| GO CC Term Nuclear Envelope | 26 (43.3%) | 18 (32.7%) | 28 (32.9%) | 32 (24.1%) | 18 (51.4%) | 18 (54.5%) |
| Samavarchi-Tehrani et al. 2018 | Viral BioID (BirA∗) Go CC term Nuclear envelope |
Flp-in HeLa 24 (40.7%) |
Viral HeLa 28 (35.9%) |
Viral BJ Fib. 18 (47.4%) |
Viral MEF 21 (48.8%) |
| May et al. 2020 (Roux lab) | LMNA BioID in A549 cells – Go CC term Nuclear envelope |
BirA∗ 26 (48.1%) |
TurboID 43 (29.1%) |
The total number of proteins identified in each BioID experiment is listed together with the number of high-confidence interactors, human orthologs, and number annotated with the GO CC terms “Nuclear Envelope.” The number of genes with this annotation from the Samavarchi-Tehrani et al. and May et al. publications are also listed.
Fig. 3.

Functional enrichment of LMNA proximal partners.A, schematic showing all the high-confidence interactors annotated with the term nuclear envelope or nuclear membrane. The color coding indicates in how many of the experiments this interactor was identified. B, overlap between all high-confidence proteins identified using both TurboID and miniTurbo by mRNA injection or transgenic expression (left), their mapped human orthologs (middle), and the human orthologs annotated as “nuclear envelope” by GO CC (right).
Comparison With Other Datasets
To further benchmark our data, we compared the human orthologs of the identified zebrafish proteins with published lamin A interactor data. The BioGRID database lists 893 (H. sapiens, accessed April 04, 2021) unique interactors for the human lamin A (LMNA) protein. These interactors were identified predominantly by yeast two-hybrid (~38%), affinity capture-MS (~25%) and proximity labeling (~25%). Across the different conditions we tested, 41 to 58% of the proximal interactors identified were already annotated in the BioGRID database (Table 3).
We next compared our data to published BioID datasets that used LMNA as a bait. We selected these because they allow us to compare specifically to datasets from our own laboratory (i.e., generated with consistent purification and spectrometric analysis protocols (51)) or using TurboID (52). Samavarchi-Tehrani et al. (51) used LMNA as a bait in their development of a lentiviral BioID toolkit and profiled LMNA in four conditions in mammalian cells. In their experiments, 18 to 28 (36–49% of the total high-confidence interactors identified) of the proteins were identified in various cell lines with a BFDR ≤1% had GO CC annotations of nuclear membrane or nuclear envelope. More recently, May et al. (52) identified 26 and 43 proteins (48 and 29% of the total high-confidence interactors identified) with these annotations in BioID experiments using BirA∗ and TurboID, respectively, in cell culture. Our results with 18 to 32 (25–49% of the total high-confidence interactors identified) filtered hits having these annotations demonstrate that our method can produce results on par with previous data (Table 3). We then compared all the high-confidence hits across these three datasets (all conditions were pooled for each dataset). Our zebrafish dataset shared 32 and 31 hits with the Samavarchi-Tehrani et al. and May et al. datasets, respectively, while these datasets shared 40 hits with each other, 22 of which were among those also in common to our zebrafish dataset (supplemental Table S5). We conclude that our zebrafish dataset identifies similar numbers and percentages of known nuclear envelope proteins as identified in previously published datasets, demonstrating that performing proximity labeling in zebrafish using our TurboID-based method is a robust method for identifying proximal interactors in vivo in a vertebrate animal model.
Parameters Influencing the Recovery of Proximal Partners in Transgenic Experiments
We next tested, using the TurboID transgenics system, the effect of reducing the concentration of biotin, omitting the heat shock, and omitting the preincubation step with biotin. Omitting biotin or using concentrations <800 μM drastically reduced labeling efficiency. Summed spectra (across two replicates) for the 54 proteins identified by TurboID-LMNA with 800 μM biotin added at 0 hpf with a BFDR ≤1% ranged from 2351 counts with 800 μM added at 0 hpf to 337 with no biotin (Fig. 4A and supplemental Table S6). Without induction of protein expression by heat shock, the total spectral counts for these 54 TurboID-LMNA high-confidence proximal interactors were further reduced (Fig. 4A and supplemental Table S6), and no protein passed the SAINT filtering threshold (Fig. 4B and supplemental Table S7). The slightly higher level of spectral counts compared with control (Fig. 4A) may be due to the expression of HSP70 in the zebrafish lens during normal development (62) that drives transgene expression for a short period even in the absence of heat shock induction; alternatively, low-level leaky expression of the transgene could explain these results (65, 66). Regardless, the low level of capture confirmed that the system is sufficiently nonleaky and that the induction timing can be controlled.
Fig. 4.

Parameters influencing the recovery of proximal partners in transgenic experiments.A, the total spectral counts from both replicates for the 54 proteins identified with a BFDR ≤1% in the 800 μM preload condition were summed for each condition and the total compared. B, the 25 most abundant proteins (by spectral counts) with a BFDR ≤1% from the earlier TurboID-LMNA experiment (Fig. 2F) are listed, alongside their abundance across conditions. C, schematic of the timeline for biotin supplementation, induction of expression, and collection of embryos.
To see whether the preloading with biotin that we had performed was necessary to enable efficient labeling, we performed an experiment in which biotin was added to the egg water only following heat shock, from 61 to 72 hpf (rather than being present from 0 to 72 hpf) (Fig. 4C). In this case, 953 proteins were identified across replicates, compared with 941 for the 0 to 72 hpf supplementation (this number is lower than earlier as two replicates were used for this comparison versus three earlier). After filtering against controls using SAINT, 56 and 54 proteins with an BFDR ≤1% remained for the 61 hpf and 0 hpf biotin supplementation samples, respectively, of which 32 overlapped (supplemental Fig. S5, A–C and supplemental Table S7). The shared proteins between these conditions were largely among the more abundant (by spectral count) high-confidence interactors, with 21 of the shared interactors being among the top 25 most abundant interactors from each condition. GO profiling of the human orthologs of the identified high-confidence interactors found 23 and 22 proteins annotated as nuclear envelope for the 0 to 72 and 61 to 72 h biotin supplementation conditions, respectively, 19 of which were shared. Some enriched categories differed between conditions, with nucleoplasm and endoplasmic reticulum annotations being enriched only in the 0 to 72 h condition, suggesting a potential increased labeling range with biotin preloading, or potentially an increased background biotinylation signal that was not well filtered out (supplemental Table S8). Importantly, however, these results indicate that omitting the preincubation with biotin still enables recovery of relevant proximal partners, albeit at a slightly lower level. These results suggest that the increased flexibility of labeling timing provided by removing the biotin preloading step makes this the better method for performing these experiments.
Discussion
In this study, we have optimized two methods, mRNA injection and transgenesis, for performing proximity-dependent TurboID labeling in zebrafish embryos.
Two recent publications have already demonstrated the use of proximity-dependent labeling in zebrafish. Xiong et al. (32) used a conditionally stabilized GFP-nanobody fused to TurboID to target proximal labeling to GFP tagged proteins in muscle, while Pronobis et al. (26) used BioID2 targeted to cardiomyocytes to investigate proteome changes during cardiac regeneration in adult fish. Both these studies showed promise for investigating proximal interactors in vivo in the zebrafish, and the Xiong et al.’s (32) manuscript is particularly interesting in that it permits use of existing GFP fusion transgenics directly after a cross with a universal GFP-nanobody expressing line. Our approach differs from these two studies in a number of ways. Firstly, we determined that both miniTurbo and TurboID are appropriate for proximity-dependent biotinylation in zebrafish. Secondly, we optimized both an mRNA injection and a transgenic system for proximity-dependent biotinylation, increasing the flexibility of the zebrafish biotinylation toolbox (discussed below). Thirdly, we performed an extensive comparison with previously published proximity labeling datasets in cell lines, making the assessment of the robustness of the methods more comprehensive. Lastly, using TurboID, we were able to perform proximity labeling in shorter labeling times (12 h) than what was published previously, which should enable better temporal studies of developmental stages.
The greatest advantage of using mRNA injection-based expression is that it allows interrogating the proximal proteome of a protein of interest in days rather than in the weeks/months required for the generation of transgenic animals. Protein expression in mRNA injected embryos is ubiquitous and robust, and we show it can be used to reliably identify proximal interactors. While the need to inject hundreds of embryos per replicate can seem daunting, we have not found this to be an obstacle, as we regularly inject 1000+ embryos in a single morning. Additionally, embryos can be frozen following deyolking and pooled from multiple injections to generate sufficient quantity for analysis. We also note that further advances in methods to purify biotinylated proteins from small amounts of input material (67) and in sensitive mass spectrometers (e.g., (68)), may further decrease the workload associated with mRNA injection. Another advantage is the ease with which protein expression levels can be varied, simply by altering the mRNA concentration injected. Some drawbacks of mRNA injection-based expression are the relatively short duration of expression, inability to control expression timing, and inability to restrict expression to specific tissues or cell types. Ultimately, whether mRNA injection is the method of choice depends on the question asked. If the experimental goal is simply to identify the proximity interactome of a protein of interest during early development, and the specific timing of the labeling is not critical, mRNA injections will be a fast and effective choice. Additionally, mRNA injections can be performed in embryos from transgenic crosses, allowing for the comparison of proximity interactomes across genetic backgrounds, such as knockouts, without the lengthy process of generating multiple new transgenic crosses for each background. In some instances, researchers may also prefer to initially perform their proximity labeling using mRNA injections, before moving on to generating stable transgenic lines. This would at the minimum provide some evidence that the tagging of the protein of interest with the enzyme does not alter its localization and permits recovery of specific biotinylated preys.
On the other hand, while generating transgenic animals requires a greater upfront investment of time, there are a number of advantages over mRNA injections. Namely, conditional expression can be achieved at any age using a range of available induction systems. Our current heat shock promoter-based approach offers researchers the opportunity to restrict expression to specific time points during development, which should enable the study of dynamic changes in proximal interactome over the course of early development. While we have not explicitly demonstrated this here, crosses with other lines can be performed to investigate proximity interactions in diverse genetic backgrounds. Additional transgenic systems could also be generated that enable tissue-specific expression by the use of tissue-specific promoters and enhancers, though this may require additional optimization (e.g., to enrich the tissues/cells labeled in order to improve the signal-to-noise ratio). The studies of Pronobis et al. (26) have demonstrated that biotin ligase-based proximity labeling is also feasible in adult zebrafish, and our heat shock promoter transgenics system should be appropriate to study proximal interactions in adults. Whether the biotin will need to be supplied through intraperitoneal injection as in Pronobis et al., or whether there will be sufficient biotin in the water to enable biotinylation by the more active TurboID enzyme will however need to be defined.
While we demonstrate that both the TurboID and miniTurbo enzymes can be used for performing BioID in the zebrafish, there were some differences. We found that in transgenic fish lines, the miniTurbo fusion constructs showed higher expression as compared with TurboID constructs. However, levels of biotinylation as well as protein recovery were higher using TurboID. The expression levels combined with the protein recovery suggest that while the smaller size of miniTurbo may result in better expression, the higher biotinylation efficiency of TurboID compensates for the lower expression. These factors should be considered when deciding which enzyme to use. When using short labeling times, such as when using inducible expression to query different timepoints during development, the higher efficiency of TurboID should allow for shorter labeling periods without significant decreases in sensitivity. On the other hand, if performing noninducible tissue-specific labeling, the higher expression levels of miniTurbo may provide equally useful results. Finally, others have found that the high efficiency of TurboID leads to toxicity when expressed constitutively and ubiquitously in flies, presumably through scavenging free biotin so efficiently (9). While we have not found this to be the case in our zebrafish experiments so far, we cannot rule it out, and it is possible that miniTurbo may be more appropriate in these cases. We also noted that our zebrafish experiments, while yielding high-quality data, were less reproducible than the cell culture experiments we have performed until now. In cell culture experiments, a skilled experimentalist should generate data that is highly similar across experiments (with correlation values easily exceeding 0.9). In the past, this has led us to elect, for cost–benefit reasons in large scale, to perform only two biological replicates per condition, and to use SAINTexpress to only report as significant proximal interactors those that are confidently scored in each of the duplicates. This strategy only induced a small loss in sensitivity (and no loss in specificity) in cultured cells. However, the lower correlation between pairwise replicates in zebrafish embryos makes this strategy undesirable. Here, we performed in most cases three biological replicate purifications and “compressed” these to two “virtual” purification using SAINTexpress (this takes, for each prey, the highest two spectral counts across the three purifications). This enables us to maintain some sensitivity for the proteins that are detected less consistently across the replicates. Strategies that involve higher number of biological replicates (from 3 to 5), and explore different statistical approaches to scoring enrichment, will be explored in future studies.
In conclusion, we have developed and benchmarked tools for reproducible proximity-dependent biotinylation in zebrafish that should be of general use to expand BioID in this important model vertebrate system. To allow the zebrafish community to easily take advantage of this system, we make available the pCS2+ mRNA expression vectors containing the TurboID and miniTurbo sequences for easy generation of mRNA coding for bait fusion proteins without the time consuming need to develop transgenic animals. We also make available Tol2Kit amenable vectors coding for TurboID and miniTurbo to enable transgenic expression of tagged proteins of interest (supplemental Fig. S6).
Data Availability
Datasets consisting of raw files and associated peak lists and results files have been deposited in ProteomeXchange through partner MassIVE (http://proteomics.ucsd.edu/ProteoSAFe/datasets.jsp) as complete submissions. Additional files include the sample description, the peptide/protein evidence, and the complete SAINTexpress output for each dataset, as well as a “README” file that describes the dataset composition and the experimental procedures associated with each submission. The different datasets generated here were submitted as independent entries.
MassIVE deposition 1: Rosenthal_Zebrafish_TurboID_P88_SAINT5425_TurboID_injections_2021
This dataset consists of 14 raw MS files and associated peak lists and result files comprising mRNA injections of the TurboID fused baits. MSV000087425, PXD026007
MassIVE deposition 2: Rosenthal_Zebrafish_TurboID_P88_SAINT5427_miniTurbo_injections_2021
This dataset consists of 13 raw MS files and associated peak lists and result files comprising mRNA injections of the miniTurbo fused baits. MSV000087428, PXD026021
MassIVE deposition 3: Rosenthal_Zebrafish_TurboID_P88_SAINT5428_TurboID_transgenics_2021
This dataset consists of nine raw MS files and associated peak lists and result files comprising transgenics (heat shock) analysis of the TurboID fused baits. MSV000087429, PXD026022
MassIVE deposition 4: Rosenthal_Zebrafish_TurboID_P88_SAINT5429_miniTurbo_transgenics_2021
This dataset consists of nine raw MS files and associated peak lists and result files comprising transgenics (heat shock) analysis of the miniTurbo fused baits. MSV000087430, PXD026023
MassIVE deposition 5: Rosenthal_Zebrafish_TurboID_P88_SAINT5454_TurboID_transgenics_conditions_2021
This dataset consists of 22 raw MS files and associated peak lists and result files comprising transgenics (heat shock) analysis of the TurboID and comparison of conditions (biotin and heat shock timing and concentration). MSV000087431, PXD026024
Supplemental data
This article contains supplemental data.
Conflicts of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgments
We thank Brett Larsen and Cassandra Wong for assistance with mass spectrometry and Payman Samavarchi-Tehrani for helpful discussion and technical advice. We also thank the SickKids Zebrafish Facility staff, Alejandro Salazar, Elyjah Schimmens, Melissa Westaway, and Scott Knox for animal husbandry. Work in the Gingras lab was supported by a Canadian Institutes of Health Research (CIHR) Foundation Grant (FDN 143301). Proteomics work was performed at the Network Biology Collaborative Centre at the Lunenfeld-Tanenbaum Research Institute, a facility supported by Canada Foundation for Innovation funding, by the Ontario Government, and by Genome Canada and Ontario Genomics (OGI-139). Work in the Scott lab was supported by a Canadian Institutes of Health Research (CIHR) project Grant (PJT 153000) and a Natural Sciences and Engineering Research Council (NSERC) Discovery Grant (RGPIN-2017-06502).
Funding and additional information
S. M. R. is supported by a SickKids Restracomp scholarship and T. C. B. was supported by Dow Graduate Research and Lester Wolfe Fellowships. A.-C. G. is the Canada Research Chair in Functional Proteomics and the Lea Reichmann Chair in Cancer Proteomics.
Author contributions
S. M. R., I. C. S., and A.-C. G. conceptualization; S. M. R. formal analysis; I. C. S. and A.-C. G. funding acquisition; S. M. R., T. M., and H. A. investigation; S. M. R., T. M., I. C. S., and A.-C. G. methodology; I. C. S. and A.-C. G. project administration; T. C. B. and A. Y. T. resources; I. C. S. and A.-C. G. supervision; A.-C. G. validation; S. M. R. visualization; S. M. R., I. C. S., and A.-C. G. writing—original draft; T. C. B. and A. Y. T. writing—review and editing.
Contributor Information
Ian C. Scott, Email: ian.scott@sickkids.ca.
Anne-Claude Gingras, Email: gingras@lunenfeld.ca.
Supplemental Data
References
- 1.Lieschke G.J., Currie P.D. Animal models of human disease: Zebrafish swim into view. Nat. Rev. Genet. 2007;8:353–367. doi: 10.1038/nrg2091. [DOI] [PubMed] [Google Scholar]
- 2.Koster R., Sassen W.A. A molecular toolbox for genetic manipulation of zebrafish. Adv. Genomics Genet. 2015;5:151. [Google Scholar]
- 3.Xin Y., Duan C. Methods in Molecular Biology. Humana Press Inc; Clifton, NJ: 2018. pp. 205–211. [Google Scholar]
- 4.Felker A., Mosimann C. Contemporary zebrafish transgenesis with Tol2 and application for Cre/lox recombination experiments. Methods Cell Biol. 2016;135:219–244. doi: 10.1016/bs.mcb.2016.01.009. [DOI] [PubMed] [Google Scholar]
- 5.Christopher J.A., Stadler C., Martin C.E., Morgenstern M., Pan Y., Betsinger C.N., Rattray D.G., Mahdessian D., Gingras A.-C., Warscheid B., Lehtiö J., Cristea I.M., Foster L.J., Emili A., Lilley K.S. Subcellular proteomics. Nat. Rev. Methods Primers. 2021;11:1–24. doi: 10.1038/s43586-021-00029-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gingras A.-C., Abe K.T., Raught B. Getting to know the neighborhood: Using proximity-dependent biotinylation to characterize protein complexes and map organelles. Curr. Opin. Chem. Biol. 2019;48:44–54. doi: 10.1016/j.cbpa.2018.10.017. [DOI] [PubMed] [Google Scholar]
- 7.Roux K.J., Kim D.I., Raida M., Burke B. A promiscuous biotin ligase fusion protein identifies proximal and interacting proteins in mammalian cells. J. Cell Biol. 2012;196:801–810. doi: 10.1083/jcb.201112098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rhee H.W., Zou P., Udeshi N.D., Martell J.D., Mootha V.K., Carr S.A., Ting A.Y. Proteomic mapping of mitochondria in living cells via spatially restricted enzymatic tagging. Science. 2013;339:1328–1331. doi: 10.1126/science.1230593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Branon T.C., Bosch J.A., Sanchez A.D., Udeshi N.D., Svinkina T., Carr S.A., Feldman J.L., Perrimon N., Ting A.Y. Efficient proximity labeling in living cells and organisms with TurboID. Nat. Biotechnol. 2018;36:880–887. doi: 10.1038/nbt.4201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kim D.I., Jensen S.C., Noble K.A., Kc B., Roux K.H., Motamedchaboki K., Roux K.J. An improved smaller biotin ligase for BioID proximity labeling. Mol. Biol. Cell. 2016;27:1188–1196. doi: 10.1091/mbc.E15-12-0844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ramanathan M., Majzoub K., Rao D.S., Neela P.H., Zarnegar B.J., Mondal S., Roth J.G., Gai H., Kovalski J.R., Siprashvili Z., Palmer T.D., Carette J.E., Khavari P.A. RNA-protein interaction detection in living cells. Nat. Methods. 2018;15:207–212. doi: 10.1038/nmeth.4601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Samavarchi-Tehrani P., Samson R., Gingras A.-C. Proximity dependent biotinylation: Key enzymes and adaptation to proteomics approaches. Mol. Cell. Proteomics. 2020;19:757–773. doi: 10.1074/mcp.R120.001941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kim D.I., Birendra K.C., Zhu W., Motamedchaboki K., Doye V., Roux K.J. Probing nuclear pore complex architecture with proximity-dependent biotinylation. Proc. Natl. Acad. Sci. U. S. A. 2014;111:E2453–E2461. doi: 10.1073/pnas.1406459111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Batsios P., Meyer I., Gräf R. Methods in Enzymology. Academic Press Inc; Clifton, NJ: 2016. pp. 23–42. [DOI] [PubMed] [Google Scholar]
- 15.Kehrer J., Frischknecht F., Mair G.R. Proteomic analysis of the plasmodium berghei gametocyte egressome and vesicular bioid of osmiophilic body proteins identifies merozoite trap-like protein (MTRAP) as an essential factor for parasite transmission. Mol. Cell. Proteomics. 2016;15:2852–2862. doi: 10.1074/mcp.M116.058263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chen A.L., Kim E.W., Toh J.Y., Vashisht A.A., Rashoff A.Q., Van C., Huang A.S., Moon A.S., Bell H.N., Bentolila L.A., Wohlschlegel J.A., Bradley P.J. Novel components of the toxoplasma inner membrane complex revealed by BioID. mBio. 2015;6 doi: 10.1128/mBio.02357-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Morriswood B., Havlicek K., Demmel L., Yavuz S., Sealey-Cardona M., Vidilaseris K., Anrather D., Kostan J., Djinovic-Carugo K., Roux K.J., Warren G. Novel bilobe components in Trypanosoma brucei identified using proximity-dependent biotinylation. Eukaryot. Cell. 2013;12:356–367. doi: 10.1128/EC.00326-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Opitz N., Schmitt K., Hofer-Pretz V., Neumann B., Krebber H., Braus G.H., Valerius O. Capturing the Asc1p/receptor for activated C kinase 1 (RACK1) microenvironment at the head region of the 40S ribosome with quantitative BioID in yeast. Mol. Cell. Proteomics. 2017;16:2199–2218. doi: 10.1074/mcp.M116.066654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Khan M., Youn J.-Y., Gingras A.-C., Subramaniam R., Desveaux D. In planta proximity dependent biotin identification (BioID) Sci. Rep. 2018;8:9212. doi: 10.1038/s41598-018-27500-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Brudvig J.J., Cain J.T., Schmidt-Grimminger G.G., Stumpo D.J., Roux K.J., Blackshear P.J., Weimer J.M. MARCKS is necessary for netrin-DCC signaling and corpus callosum formation. Mol. Neurobiol. 2018;55:8388–8402. doi: 10.1007/s12035-018-0990-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rudolph F., Fink C., Hüttemeister J., Kirchner M., Radke M.H., Lopez Carballo J., Wagner E., Kohl T., Lehnart S.E., Mertins P., Gotthardt M. Deconstructing sarcomeric structure–function relations in titin-BioID knock-in mice. Nat. Commun. 2020;11:3133. doi: 10.1038/s41467-020-16929-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Feng W., Liu C., Spinozzi S., Wang L., Evans S.M., Chen J. Identifying the cardiac dyad proteome in vivo by a BioID2 knock-in strategy. Circulation. 2020;141:940–942. doi: 10.1161/CIRCULATIONAHA.119.043434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Uezu A., Kanak D.J., Bradshaw T.W.A., Soderblom E.J., Catavero C.M., Burette A.C., Weinberg R.J., Soderling S.H. Identification of an elaborate complex mediating postsynaptic inhibition. Science. 2016;353:1123–1129. doi: 10.1126/science.aag0821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Spence E.F., Dube S., Uezu A., Locke M., Soderblom E.J., Soderling S.H. In vivo proximity proteomics of nascent synapses reveals a novel regulator of cytoskeleton-mediated synaptic maturation. Nat. Commun. 2019;10:386. doi: 10.1038/s41467-019-08288-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Dingar D., Kalkat M., Chan P.-K., Srikumar T., Bailey S.D., Tu W.B., Coyaud E., Ponzielli R., Kolyar M., Jurisica I., Huang A., Lupien M., Penn L.Z., Raught B. BioID identifies novel c-MYC interacting partners in cultured cells and xenograft tumors. J. Proteomics. 2015;118:95–111. doi: 10.1016/j.jprot.2014.09.029. [DOI] [PubMed] [Google Scholar]
- 26.Pronobis M.I., Zheng S., Singh S.P., Goldman J.A., Poss K.D. In vivo proximity labeling identifies cardiomyocyte protein networks during zebrafish heart regeneration. Elife. 2021;10 doi: 10.7554/eLife.66079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhang B., Zhang Y., Liu J.-L. Highly effective proximate labeling in Drosophila. G3 (Bethesda) 2021;11 doi: 10.1093/g3journal/jkab077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mair A., Xu S.L., Branon T.C., Ting A.Y., Bergmann D.C. Proximity labeling of protein complexes and cell type specific organellar proteomes in Arabidopsis enabled by TurboID. Elife. 2019;8 doi: 10.7554/eLife.47864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhang Y., Song G., Lal N.K., Nagalakshmi U., Li Y., Zheng W., Huang P.-J., Branon T.C., Ting A.Y., Walley J.W., Dinesh-Kumar S.P. TurboID-based proximity labeling reveals that UBR7 is a regulator of N NLR immune receptor-mediated immunity. Nat. Commun. 2019;10:3252. doi: 10.1038/s41467-019-11202-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Larochelle M., Bergeron D., Arcand B., Bachand F. Proximity-dependent biotinylation mediated by TurboID to identify protein-protein interaction networks in yeast. J. Cell Sci. 2019;132 doi: 10.1242/jcs.232249. [DOI] [PubMed] [Google Scholar]
- 31.Takano T., Wallace J.T., Baldwin K.T., Purkey A.M., Uezu A., Courtland J.L., Soderblom E.J., Shimogori T., Maness P.F., Eroglu C., Soderling S.H. Chemico-genetic discovery of astrocytic control of inhibition in vivo. Nature. 2020;588:296–302. doi: 10.1038/s41586-020-2926-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Xiong Z., Lo H.P., McMahon K.A., Martel N., Jones A., Hill M.M., Parton R.G., Hall T.E. In vivo proteomic mapping through gfp-directed proximity-dependent biotin labelling in zebrafish. Elife. 2021;10 doi: 10.7554/eLife.64631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Grzegorski S.J., Chiari E.F., Robbins A., Kish P.E., Kahana A. Natural variability of Kozak sequences correlates with function in a zebrafish model. PLoS One. 2014;9 doi: 10.1371/journal.pone.0108475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kwan K.M., Fujimoto E., Grabher C., Mangum B.D., Hardy M.E., Campbell D.S., Parant J.M., Yost H.J., Kanki J.P., Chien C. Bin. The Tol2kit: A multisite gateway-based construction kit for Tol2 transposon transgenesis constructs. Dev. Dyn. 2007;236:3088–3099. doi: 10.1002/dvdy.21343. [DOI] [PubMed] [Google Scholar]
- 35.Le X., Langenau D.M., Keefe M.D., Kutok J.L., Neuberg D.S., Zon L.I. Heat shock-inducible Cre/Lox approaches to induce diverse types of tumors and hyperplasia in transgenic zebrafish. Proc. Natl. Acad. Sci. U. S. A. 2007;104:9410–9415. doi: 10.1073/pnas.0611302104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ádám A., Bártfai R., Lele Z., Krone P.H., Orbán L. Heat-inducible expression of a reporter gene detected by transient assay in zebrafish. Exp. Cell Res. 2000;256:282–290. doi: 10.1006/excr.2000.4805. [DOI] [PubMed] [Google Scholar]
- 37.Scheer N., Riedl I., Warren J.T., Kuwada J.Y., Campos-Ortega J.A. A quantitative analysis of the kinetics of Gal4 activator and effector gene expression in the zebrafish. Mech. Dev. 2002;112:9–14. doi: 10.1016/s0925-4773(01)00621-9. [DOI] [PubMed] [Google Scholar]
- 38.Shen M.-C., Ozacar A.T., Osgood M., Boeras C., Pink J., Thomas J., Kohtz J.D., Karlstrom R. Heat-shock-mediated conditional regulation of hedgehog/gli signaling in zebrafish. Dev. Dyn. 2013;242:539–549. doi: 10.1002/dvdy.23955. [DOI] [PubMed] [Google Scholar]
- 39.Link V., Shevchenko A., Heisenberg C.P. Proteomics of early zebrafish embryos. BMC Dev. Biol. 2006;6:1. doi: 10.1186/1471-213X-6-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Santos D., Monteiro S.M., Luzio A. Methods in Molecular Biology. Humana Press Inc; Clifton, NJ: 2018. pp. 365–371. [DOI] [PubMed] [Google Scholar]
- 41.Hesketh G.G., Youn J.Y., Samavarchi-Tehrani P., Raught B., Gingras A.C. Methods in Molecular Biology. Humana Press Inc; Clifton, NJ: 2017. pp. 115–136. [DOI] [PubMed] [Google Scholar]
- 42.Liu G., Zhang J., Larsen B., Stark C., Breitkreutz A., Lin Z.Y., Breitkreutz B.J., Ding Y., Colwill K., Pasculescu A., Pawson T., Wrana J.L., Nesvizhskii A.I., Raught B., Tyers M. ProHits: Integrated software for mass spectrometry-based interaction proteomics. Nat. Biotechnol. 2010;28:1015–1017. doi: 10.1038/nbt1010-1015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kessner D., Chambers M., Burke R., Agus D., Mallick P. ProteoWizard: Open source software for rapid proteomics tools development. Bioinformatics. 2008;24:2534–2536. doi: 10.1093/bioinformatics/btn323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Perkins D.N., Pappin D.J.C., Creasy D.M., Cottrell J.S. Electrophoresis. Wiley-VCH Verlag; Weinheim, Germany: 1999. pp. 3551–3567. [DOI] [PubMed] [Google Scholar]
- 45.Eng J.K., Jahan T.A., Hoopmann M.R. Comet: An open-source MS/MS sequence database search tool. Proteomics. 2013;13:22–24. doi: 10.1002/pmic.201200439. [DOI] [PubMed] [Google Scholar]
- 46.Deutsch E.W., Mendoza L., Shteynberg D., Farrah T., Lam H., Tasman N., Sun Z., Nilsson E., Pratt B., Prazen B., Eng J.K., Martin D.B., Nesvizhskii A.I., Aebersold R. A guided tour of the trans-proteomic pipeline. Proteomics. 2010;10:1150–1159. doi: 10.1002/pmic.200900375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Shteynberg D., Deutsch E.W., Lam H., Eng J.K., Sun Z., Tasman N., Mendoza L., Moritz R.L., Aebersold R., Nesvizhskii A.I. iProphet: Multi-level integrative analysis of shotgun proteomic data improves peptide and protein identification rates and error estimates. Mol. Cell. Proteomics. 2011;10 doi: 10.1074/mcp.M111.007690. M111.007690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Teo G., Liu G., Zhang J., Nesvizhskii A.I., Gingras A.C., Choi H. SAINTexpress: Improvements and additional features in significance analysis of INTeractome software. J. Proteomics. 2014;100:37–43. doi: 10.1016/j.jprot.2013.10.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Reimand J., Arak T., Adler P., Kolberg L., Reisberg S., Peterson H., Vilo J. g:Profiler-a web server for functional interpretation of gene lists (2016 update) Nucleic Acids Res. 2016;44:W83–W89. doi: 10.1093/nar/gkw199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Agarwala R., Barrett T., Beck J., Benson D.A., Bollin C., Bolton E., Bourexis D., Brister J.R., Bryant S.H., Canese K., Cavanaugh M., Charowhas C., Clark K., Dondoshansky I., Feolo M. Database resources of the National Center for Biotechnology Information. Nucleic Acids Res. 2018;46:D8–D13. doi: 10.1093/nar/gkx1095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Samavarchi-Tehrani P., Abdouni H., Samson R., Gingras A.C. A versatile lentiviral delivery toolkit for proximitydependent biotinylation in diverse cell types authors. Mol. Cell. Proteomics. 2018;17:2256–2269. doi: 10.1074/mcp.TIR118.000902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.May D.G., Scott K.L., Campos A.R., Roux K.J. Comparative application of BioID and TurboID for protein-proximity biotinylation. Cells. 2020;9:1070. doi: 10.3390/cells9051070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Knight J.D.R., Choi H., Gupta G.D., Pelletier L., Raught B., Nesvizhskii A.I., Gingras A.C. ProHits-viz: A suite of web tools for visualizing interaction proteomics data. Nat. Methods. 2017;14:645–646. doi: 10.1038/nmeth.4330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Verma A.D., Parnaik V.K. Heart-specific expression of laminopathic mutations in transgenic zebrafish. Cell Biol. Int. 2017;41:809–819. doi: 10.1002/cbin.10784. [DOI] [PubMed] [Google Scholar]
- 55.Manju K., Muralikrishna B., Parnaik V.K. Expression of disease-causing lamin A mutants impairs the formation of DNA repair foci. J. Cell Sci. 2006;119:2704–2714. doi: 10.1242/jcs.03009. [DOI] [PubMed] [Google Scholar]
- 56.Ahmed R., Spikings E., Zhou S., Thompsett A., Zhang T. Pre-hybridisation: An efficient way of suppressing endogenous biotin-binding activity inherent to biotin-streptavidin detection system. J. Immunol. Methods. 2014;406:143–147. doi: 10.1016/j.jim.2014.03.010. [DOI] [PubMed] [Google Scholar]
- 57.Grant M.K.O., Shapiro S.L., Ashe K.H., Liu P., Zahs K.R. A cautionary tale: Endogenous biotinylated proteins and exogenously-introduced protein A cause antibody-independent artefacts in western blot studies of brain-derived proteins. Biol. Proced. Online. 2019;21:6. doi: 10.1186/s12575-019-0095-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Sanchez A.D., Branon T., Cote L., Papagiannakis A., Liang X., Pickett M., Shen K., Jacobs-Wagner C., Ting A., Feldman J. Proximity labeling reveals non-centrosomal microtubule-organizing center components required for microtubule growth and localization. Curr. Biol. 2020 doi: 10.1016/j.cub.2021.06.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kimmel C.B., Ballard W.W., Kimmel S.R., Ullmann B., Schilling T.F. Stages of embryonic development of the zebrafish. Dev. Dyn. 1995;203:253–310. doi: 10.1002/aja.1002030302. [DOI] [PubMed] [Google Scholar]
- 60.Mellacheruvu D., Wright Z., Couzens A.L., Lambert J.P., St-Denis N.A., Li T., Miteva Y.V., Hauri S., Sardiu M.E., Low T.Y., Halim V.A., Bagshaw R.D., Hubner N.C., Al-Hakim A., Bouchard A. The CRAPome: A contaminant repository for affinity purification-mass spectrometry data. Nat. Methods. 2013;10:730–736. doi: 10.1038/nmeth.2557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Shoji W., Sato-Maeda M. Application of heat shock promoter in transgenic zebrafish. Dev. Growth Differ. 2008;50:401–406. doi: 10.1111/j.1440-169X.2008.01038.x. [DOI] [PubMed] [Google Scholar]
- 62.Blechinger S.R., Evans T.G., Tang P.T., Kuwada J.Y., Warren J.T., Krone P.H. The heat-inducible zebrafish hsp70 gene is expressed during normal lens development under non-stress conditions. Mech. Dev. 2002;112:213–215. doi: 10.1016/s0925-4773(01)00652-9. [DOI] [PubMed] [Google Scholar]
- 63.Brox S., Ritter A.P., Küster E., Reemtsma T. A quantitative HPLC-MS/MS method for studying internal concentrations and toxicokinetics of 34 polar analytes in zebrafish (Danio rerio) embryos. Anal. Bioanal. Chem. 2014;406:4831–4840. doi: 10.1007/s00216-014-7929-y. [DOI] [PubMed] [Google Scholar]
- 64.Go C.D., Knight J.D.R., Rajasekharan A., Rathod B., Hesketh G.G., Abe K.T., Youn J.-Y., Samavarchi-Tehrani P., Zhang H., Zhu L.Y., Popiel E., Lambert J.-P., Coyaud, É., Cheung S.W.T., Rajendran D., Wong C.J. A proximity-dependent biotinylation map of a human cell. Nature. 2021;595:120–124. doi: 10.1038/s41586-021-03592-2. [DOI] [PubMed] [Google Scholar]
- 65.Yeh F., Hsu T. Differential regulation of spontaneous and heat-induced HSP 70 expression in developing zebrafish (Danio rerio) J. Exp. Zool. 2002;293:349–359. doi: 10.1002/jez.10093. [DOI] [PubMed] [Google Scholar]
- 66.Fu-Lung Y., Todd H. Detection of a spontaneous high expression of heat shock protein 70 in developing zebrafish (Danio rerio) Biotechnol. Biochem. 2000;64:592–595. doi: 10.1271/bbb.64.592. [DOI] [PubMed] [Google Scholar]
- 67.Furlan C., Dirks R.A.M., Thomas P.C., Jones R.C., Wang J., Lynch M., Marks H., Vermeulen M. Miniaturised interaction proteomics on a microfluidic platform with ultra-low input requirements. Nat. Commun. 2019;10:1525. doi: 10.1038/s41467-019-09533-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Meier F., Brunner A.D., Koch S., Koch H., Lubeck M., Krause M., Goedecke N., Decker J., Kosinski T., Park M.A., Bache N., Hoerning O., Cox J., Räther O., Mann M. Online parallel accumulation–serial fragmentation (PASEF) with a novel trapped ion mobility mass spectrometer. Mol. Cell. Proteomics. 2018;17:2534–2545. doi: 10.1074/mcp.TIR118.000900. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Datasets consisting of raw files and associated peak lists and results files have been deposited in ProteomeXchange through partner MassIVE (http://proteomics.ucsd.edu/ProteoSAFe/datasets.jsp) as complete submissions. Additional files include the sample description, the peptide/protein evidence, and the complete SAINTexpress output for each dataset, as well as a “README” file that describes the dataset composition and the experimental procedures associated with each submission. The different datasets generated here were submitted as independent entries.
MassIVE deposition 1: Rosenthal_Zebrafish_TurboID_P88_SAINT5425_TurboID_injections_2021
This dataset consists of 14 raw MS files and associated peak lists and result files comprising mRNA injections of the TurboID fused baits. MSV000087425, PXD026007
MassIVE deposition 2: Rosenthal_Zebrafish_TurboID_P88_SAINT5427_miniTurbo_injections_2021
This dataset consists of 13 raw MS files and associated peak lists and result files comprising mRNA injections of the miniTurbo fused baits. MSV000087428, PXD026021
MassIVE deposition 3: Rosenthal_Zebrafish_TurboID_P88_SAINT5428_TurboID_transgenics_2021
This dataset consists of nine raw MS files and associated peak lists and result files comprising transgenics (heat shock) analysis of the TurboID fused baits. MSV000087429, PXD026022
MassIVE deposition 4: Rosenthal_Zebrafish_TurboID_P88_SAINT5429_miniTurbo_transgenics_2021
This dataset consists of nine raw MS files and associated peak lists and result files comprising transgenics (heat shock) analysis of the miniTurbo fused baits. MSV000087430, PXD026023
MassIVE deposition 5: Rosenthal_Zebrafish_TurboID_P88_SAINT5454_TurboID_transgenics_conditions_2021
This dataset consists of 22 raw MS files and associated peak lists and result files comprising transgenics (heat shock) analysis of the TurboID and comparison of conditions (biotin and heat shock timing and concentration). MSV000087431, PXD026024