

Abstract
Genetic variants in components of the protein kinase A (PKA) enzyme have been associated with various defects and neoplasms in the context of Carney complex (CNC) and in isolated cases, such as in primary pigmented nodular adrenocortical disease (PPNAD), cortisol-producing adrenal adenomas (CPAs), and various cancers. PRKAR1A mutations have been found in subjects with impaired cAMP-dependent signaling and skeletal defects; bone tumors also develop in both humans and mice with Prkar1a abnormalities. We studied the PRKACB gene in 148 subjects with PPNAD and related disorders, who did not have other PKA-related defects and identified two subjects with possibly pathogenic PRKACB gene variants and unusual bone and endocrine phenotypes. The first presented with bone and other abnormalities, and carried a de novo c.858_860GAA (p.K286del) variant. The second subject carried the c.899 C>T (p.T300M) variant and had a PPNAD-like phenotype. Both variants are highly conserved in the PRKACB gene. In functional studies, the p.K286del variant affected PRKACB protein stability and led to increased PKA signaling. The p.T300M variant did not affect protein stability or response to cAMP and its pathogenicity remains uncertain. We conclude that PRKACB germline variants are uncommon but may be associated with phenotypes that resemble those of other PKA-related defects. However, detailed investigation of each variant is needed as PRKACB appears to be only rarely affected in these conditions, and variants such as p.T300M maybe proven to be clinically insignificant, whereas others (such as p.K286del) are clearly pathogenic and may lead to a novel skeletal syndrome phenotype.
Keywords: cyclic AMP, cortisol, adrenocortical hyperplasia, PRKAR1A, PRKACA, PRKACB
INTRODUCTION
Protein kinase A (PKA) is a tetrameric enzyme composed of a regulatory (R) subunit dimer and two catalytic (C) subunits. The R- and C- subunits disassociate when cyclic adenosine monophosphate (cAMP) molecules bind to each R-subunit, releasing the now-active C-subunits to phosphorylate downstream targets (Tasken and Aandahl 2004; Taylor, et al. 2012). There are four different R-subunits (RIα, RIβ, RIIα and RIIβ) and four C-subunits (Cα, Cβ, and Cγ, and PRKX) that act as serine threonine kinases; each is coded by a specific gene (PRKAR1A, PRKAR1B, PRKAR2A, PRKAR2B, PRKACA, PRKACB, PRKACG, and PRKAX, respectively) (Soberg, et al. 2013; Taylor, et al. 2012). RIα, Cα and PRKX are ubiquitously expressed, whereas the other subunits have more limited, tissue-specific distribution (Calebiro, et al. 2017; Liu, et al. 2016; Soberg, et al. 2013).
Mutations that result in activation of PKA signaling are a cause of several uncommon but significant human diseases. Somatic mutations in GNAS that lead to constitutive activation of Gαs increase production of cAMP, leading to either the complete McCune-Albright syndrome (MAS) or a form fruste consisting of only one component in isolation (Turan and Bastepe 2015), can also occur in other endocrine tumors such as GH secreting adenomas and thyroid tumors. The identification of heterozygous germline PRKAR1A mutations in patients with Carney complex (CNC) and acrodysostosis (ACRDYS1) provided further evidence that dysregulated PKA signaling was an important cause of endocrine and non-endocrine disorders (Horvath, et al. 2010; Kirschner, et al. 2000). Somatic PRKAR1A mutations are present in isolated primary pigmented nodular adrenocortical disease (PPNAD) and cortisol-producing adenomas (CPA) causing Cushing syndrome (CS) (Bertherat, et al. 2003; Bertherat, et al. 2009). PRKACA mutations are the most frequent genetic alteration in isolated CPAs (Beuschlein, et al. 2014; Di Dalmazi, et al. 2014). These mutations increase PRKACA basal activity via interfering with its binding to R-subunits (Calebiro, et al. 2014). A single subject with a CNC-like condition, including a growth-hormone (GH)-secreting pituitary adenoma and myxomas, along with a mild form of skeletal defects was found to have somatic mosaicism for a copy number gain of the PRKACB gene (Forlino, et al. 2014). The association of bone abnormalities with abnormal PKA function was first suggested in MAS and the discovery of osteochondromyxomas, a mixed osteo-cartilaginous tumor in subjects with CNC (Carney, et al. 2001) and the Prkar1a-haploinsufficient mice (Liu, et al. 2016; Molyneux, et al. 2010; Tsang, et al. 2010). Subjects with ACRDYS1 have specific PRKAR1A mutations (Linglart, et al. 2011). Subjects with 17q22-24 deletions that include the PRKAR1A gene also were described with a variety of skeletal abnormalities (Salpea, et al. 2014). PRKACA defects were identified in isolated PPNAD, CPAs and other adrenocortical tumors (Beuschlein, et al. 2014; Carney, et al. 2015; Di Dalmazi, et al. 2014; Lodish, et al. 2015) and cardiac myxomas (Stratakis 2018; Tseng, et al. 2017), as well as a variety of other neoplasms, including fibrolamellar hepatocellular carcinoma (Honeyman, et al. 2014) and intraductal oncocytic papillary neoplasms of the pancreas and bile duct (Singhi, et al. 2020).
Recently, a single somatic PRKACB sequencing variant was found in a CPA of a subject with CS; we fully investigated the p.S54L defect for its effect on the PRKACB protein and PKA signaling (Espiard, et al. 2018). In the present study, we report the clinical and molecular investigations of two additional genetic variants of the PRKACB gene that we identified in the germline of two subjects out of a total of 148 investigated for PKA defects. These subjects, their phenotypes, and the work described here on their PRKACB variants, add to our understanding of the pathophysiology of PKA signaling and its possible involvement in adrenal function and bone development.
MATERIALS and METHODS
Genetic studies
Subjects included in this study are part of a cohort (n = 148) recruited under the protocol 95CH0059 approved by National Institutes of Health (NIH) intramural institutional review boards for the study of PPNAD and related conditions; the parents and subjects provided consent and/or assent for the studies described here, as appropriate. The 148 subjects studied here did not have any other variants in PKA subunits (PRKAR1A, PRKAR2B, PRKACA) or related genes (data not shown). Their PRKACB sequencing data revealed either normal sequence or a few mostly previously described or non-coding variants (Supplementary Table 1). Two subjects, however, had two PRKACB variants that at least one in silico tool identified as likely pathogenic and are the subjects of this study.
The subject shown in Figure 1 (Subject #1) was consented at the Children’s Hospital of Philadelphia (CHOP). Adrenal tumor samples were obtained from subject #2 at surgery, as previously described (Courcoutsakis, et al. 2004). DNA was extracted from peripheral blood for subjects #1 and #2 and fresh-frozen tissues for subject #2, as previously described (Bertherat, et al. 2003; Espiard, et al. 2018; Horvath, et al. 2010). Comparative genomic hybridization (CGH) and whole exome sequencing (WES) were conducted as we have described elsewhere (Bertherat, et al. 2003; Espiard, et al. 2018; Horvath, et al. 2010). In brief, DNA was captured using the SureSelectXT Human All Exon version 5 Kit (Agilent, Santa Clara, USA), following the manufacturer’s protocol, and then sequenced on a paired-end mode of HiSeq2500 (Espiard, et al. 2018). The mean coverage depth ranges from 52-fold to 109-fold for the subject #1 and her parents. Her unaffected sibling was sequenced to 41-fold. Subject’s #2 germline and somatic DNA was sequenced to 108-fold. The PRKACB-coding and the flanking intronic sequences were amplified by PCR using DNA from tumor tissue and/or peripheral blood monocytes. Both strands of the amplified products were directly sequenced with forward and reverse primers, as we described previously (Espiard, et al. 2018).
Figure 1. Clinical and histological features of the subjects.
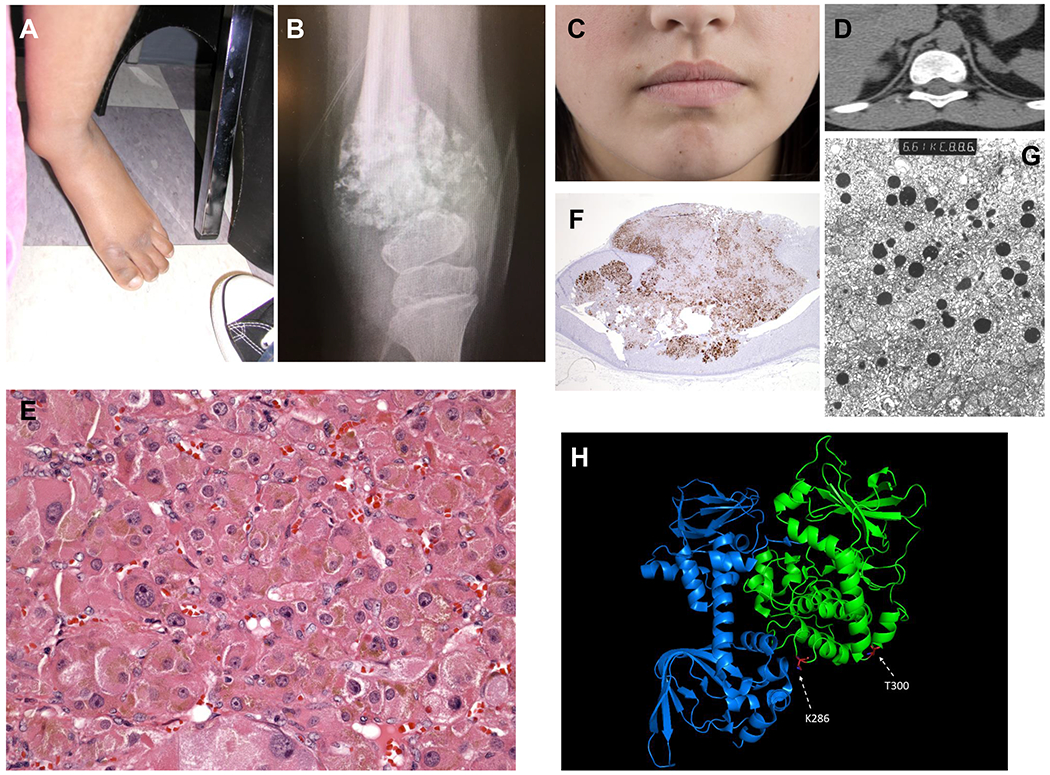
A. Left lower extremity of subject #1; B. X-Ray of the left knee joint of subject #1 shows diaphyseal widening and multiple lytic lesions; C. Subject #2 as she is today at the age of 25 years, with a few skin pigmented lesions but no perioral or mucosal spots that would be characteristic of Carney complex; D. Computed tomography shows that the subject #2 had bilateral micronodular adrenocortical hyperplasia, when she was first diagnosed with Cushing syndrome; E. Hematoxylin and eosin staining shows the characteristic eosinophilic cells that are cortisol-producing and are loaded with lipofuscin (shown as brown pigment in the cytoplasm of most cells in this section), as is the case in PPNAD and PPNAD-like lesions; F. Synaptophysin staining of her adrenal gland shows extensive staining of the cortex as it is the case for most micronodular adrenocortical hyperplasias causing ACTH-independent CS; G. Electron microscopy shows hyperplastic cortical cells with multiple lipid droplets; and H. Crystal structure of the PKA holoenzyme complex (pdb: 6NO7). In green: catalytic subunit (Cα); in blue: regulatory subunit (RIα). Location of the p.T300M and p.K286del PRKACB variants are depicted in red. pK286del is located in a position where it could interfere with binding to the regulatory subunits.
In silico modeling
Genetic variants were evaluated by MutationTaster (http://www.mutationtaster.org/), Polymorphism Phenotyping v2 algorithm tool (PolyPhen-2) (http://genetics.bwh.harvard.edu/pph2) and SIFT (Sorting Tolerant From Intolerant) algorithm (http://sift.jcvi.org) to predict the possible impact of the amino acid substitution on the structure and function of the corresponding proteins.
TCGA database
Somatic variants of PRKACB gene were extracted from the TCGA database in respect to different tissues (https://www.cancer.gov/tcga).
cDNA constructs, cell culture and protein stability
The PRKACB cDNA sequence (NM_002731.3) was inserted into expression vector, and the amino acid changes (p.S54L, p.K286del, and p.T300M) were introduced by site-directed mutagenesis as we described elsewhere (Espiard, et al. 2018). All nucleotide sequences were confirmed by Sanger sequencing. For the protein stability assay, HEK293 cells were transfected with 500ng of PRKACB vector. Cells were treated with different concentrations of proteasome inhibitors MG-132 (Milipore Sigma, St. Louis, USA), NN-DNJ (Milipore Sigma, St. Louis, USA) and Leupeptin (Milipore Sigma, St. Louis, USA) as previously described (Patronas, et al. 2012). Then HEK293 cells were transfected with the same amount of PRKACB and PRKAR1A vector. For the PKA catalytic analysis, since expression of the p.S54L and p.K286del recombinant PRKACB proteins was lower in transfected HEK293 cells than either the wild-type (WT) and the p.T300M proteins, we transfected HEK293 cells with increased amounts of the p.S54L and p.K286del cDNAs (330 ng for these two mutants and 170 ng for WT and p.T300M vectors) while keeping the regulatory subunits, PRKAR1A and PRKAR2B, constant (670 ng) to obtain similar expression levels for all PRKACB proteins (Supplementary Figure 1).
In vitro PKA activity assay with cell lysates
All experiments were performed 48h after transfection. HEK293 cells were washed twice with PBS at room temperature, scraped from the plate, and re-suspended in 300μl lysis buffer (5 mM Tris-HCl, 2 mM EDTA, pH 7.4). The cells were lysed using an Ultraturrax for 20 s on ice. Then the samples were centrifuged at 50,000 x g for 30 min at 4 °C to remove membranes. To check for equal input of catalytic subunit for the PKA activity assay, PKA subunits FLAG-Cβ, RIα and RIIβ expressions in cell lysates were determined by Western blot with specific antibodies (anti-FLAG (1:4000), #F7425, (Milipore Sigma, St. Louis, USA); anti-PKA RIα (1:1000), #610609, (BD Transduction Laboratories, San Jose, USA); anti-PKA RIIβ (1:1000), #610625, (BD Transduction Laboratories, San Jose, USA). PKA catalytic activity was measured with or without the addition of cAMP using the PepTag non-radioactive cAMP-dependent protein kinase assay (#V5340, Promega, Madison, USA) using Kemptide (LRRASLG), according to the manufacture’s instructions. Images of the gels were acquired with a gel documentation system (Herolab) and analyzed using ImageJ software (http://rsbweb.nih.gov/ij).
Immunohistochemistry (IHC)
IHC was performed on serial sections from paraffin-embedded adrenal tissue from subject #2 and control tissues. Antibody concentrations, other information and related methods are described elsewhere (Courcoutsakis, et al. 2004; Espiard, et al. 2018).
Statistical analyses
All data are presented as the mean +/− standard deviation (SD), and significance was determined by unpaired two-tailed t-test or two-way ANOVA. Statistical significance was set at P<0.05. Analyses were carried out using the GraphPadPrism 6 (GraphPad®) software.
RESULTS
Clinical case descriptions
Subject #1
A 15-year-old Hispanic female was evaluated at CHOP for extreme short stature, multiple skeletal developmental malformations, and severe developmental delay. She had been initially evaluated as an infant for hypercalcemia that was characterized by elevated or normal serum concentrations of 1,25-dihydroxyvitamin D, low or suppressed levels of serum PTH, and normal serum levels of alkaline phosphatase and phosphorus; there was mild hypercalciuria. She was successfully managed with a low calcium, low vitamin D diet, and as she matured, she was transitioned to a regular diet. A recent abdominal CT disclosed right renal cysts and a non-obstructing left renal calculus. Her skeletal abnormalities included symmetrical metaphyseal irregularities and hypoplasia of distal phalanges, cleidocranial dysostosis, bilateral coxa vara, unossified pubic ramus and history of delayed closure of the anterior fontanel, sacral hemidysplasia with absence of the right distal sacrum and a spinal cord syrinx, progressive contractures of her large joints, and displaced left ankle (Figure 1A). On examination she also had macrocephaly, triangular facies, hypertelorism and a prominent nose, and 2,34 syndactyly and shortened 4th and 5th metacarpals. She was non-verbal and non-ambulatory. Her bone age was consistently delayed relative to her chronologic age and she had normal hormonal studies; skeletal survey showed multiple features of metaphyseal chondrodysplasia with acroosteolysis of the distal upper and lower extremities (Figure 1B). The parents, an older brother, and younger sister were all unaffected.
Subject #2
We initially described this subject, seen at NIH, in 2004 when she was almost 9 years old (Courcoutsakis, et al. 2004); she is now 25 years-old (Figure 1C). She had presented with corticotropin-independent CS due to bilateral micronodular adrenocortical hyperplasia on imaging (Figure 1D) and no other tumors (Courcoutsakis, et al. 2004). She underwent bilateral adrenalectomy and the histologic features of the lesion were unusual with the nodules containing some pigment and staining positive for synaptophysin (like in PPNAD) (Figure 1E and 1F, respectively), demonstrating myeloid metaplasia (Figure 1G), and very uniformly atrophic extra-nodular cortex (Courcoutsakis, et al. 2004). We have followed this subject to the present: she remains healthy on glucocorticoid and mineralocorticoid replacement. To date, she has not developed any signs of CNC neither clinical nor biochemical despite annual surveillance with targeted imaging studies. Note that the father of subject #2 is Italian-American and her mother is mixed Irish/Italian American.
Sequencing and in silico analyses
PRKACB variants were identified by sequencing a total of 148 subjects with PPNAD or other PKA-related disorders who did not have other PKA subunit defects (Supplementary Table 1).
For subject #1, WES revealed a germline heterozygous de novo (both parents and siblings were negative) variant, NM_002731.3 c.858_860delGAA (p.K286del), in the PRKACB gene (Supplementary Table 1). The variant was confirmed by Sanger sequencing and the parents’ DNA was also sequenced (Supplementary Figure 2). SNP-microarray was 46 XX. WES showed in subject #2 a germline missense PRKACB variant, c.899C>T; p.T300M (Supplementary Table 1) - also known as p.T347M in another isoform (NM_182948.4) of the PRKACB gene (Supplementary Table 2). The variant was confirmed by Sanger sequencing (Supplementary Figure 3). DNA of the relatives from subject #2 was also sequenced and showed the presence of the variant in the unaffected father (data not shown). There were no other variants in subject’s WES that may explain the phenotype (Supplementary Table 2); subject #2 has also been screened over the years by targeted sequencing (including testing for deletions and/or copy number variants) for all the genes that have been identified and predispose to CS and bilateral adrenocortical hyperplasias, including PRKAR1A, PDE11A, PDE8B, ARMC5, and PRKACA; she carried no other variants or genomic defects in these genes. Sequencing of somatic DNA (that was extracted from her adrenal hyperplasia) of subject #2 showed no additional variants in the PRKACB gene or loss of heterozygosity at the gene locus (Supplementary Figure 3). In addition, no other abnormalities that could explain the disease were found by chromosomal and copy number variant analyses (data not shown).
Both the p.K286del and p.T300M variants are in sequences of the PRKACB protein that are highly conserved evolutionarily, similar to the p.S54L mutation that we previously described (Espiard, et al. 2018) (Supplementary Table 3) . The p.K286del is in a location of the protein where it could interfere with regulatory subunit interaction; the location of p.T300M, however, may not affect this function (Soberg, et al. 2013; Taylor, et al. 2012) (Figure 1H). The p.K286del change is rare variant (rs746464606 with minor allele frequency (MAF) of 0.000003989): there appears to be only one other carrier of an unknown phenotype in the gnomAD database (http://gnomad.broadinstitute.org/variant/1-84679925-AAAG-A). The p.T300M is also relatively uncommon, having a registered SNP (rs143624941), and MAF of 0.0003 in the European American population of the same ancestry as our patient; it was predicted to be damaging by Mutation Tester (probability: 1) and SIFT (score: 0.04), whereas Polyphen2 predicted it to be benign. Apparently, the same variant has a somewhat higher MAF in south Asians (data not shown). At the somatic level, the variant p.T300M was found in one subject with bronchus and lung cancer, and in another one with corpus uteri cancer, according to the TCGA database (Supplementary Figure 4).
In vitro expression studies
HEK293 cells that had been transfected with the p.K286del variant showed reduced expression relative to WT PRKACB protein both at 24h and 48h, similar to p.S54L. By contrast, expression levels of the p.T300M variant were unchanged at both time points (Figure 2A). Inhibition of proteasomal (PS-431 and MG-132) and lysosomal degradation (leupeptin and chloroquine) did not increase expression of the p.S54L and p.K286del mutants (Figure 2B and 2C show the MG-132 and leupeptin experiments, respectively). On the other hand, addition of the NN-DNJ chaperone molecule (Figure 2D) led to an increase in expression of the p.K286del and a lesser increase for the p.S54L protein (Figure 2E). Co-transfection with PRKAR1A subunits stabilized the p.K286del mutant protein as chaperone molecule did (Figure 2F). There was no effect of the p.T300M variant on the amount of PRKACB protein present after in vitro expression.
Figure 2. Protein stability assay in HEK293 cells.
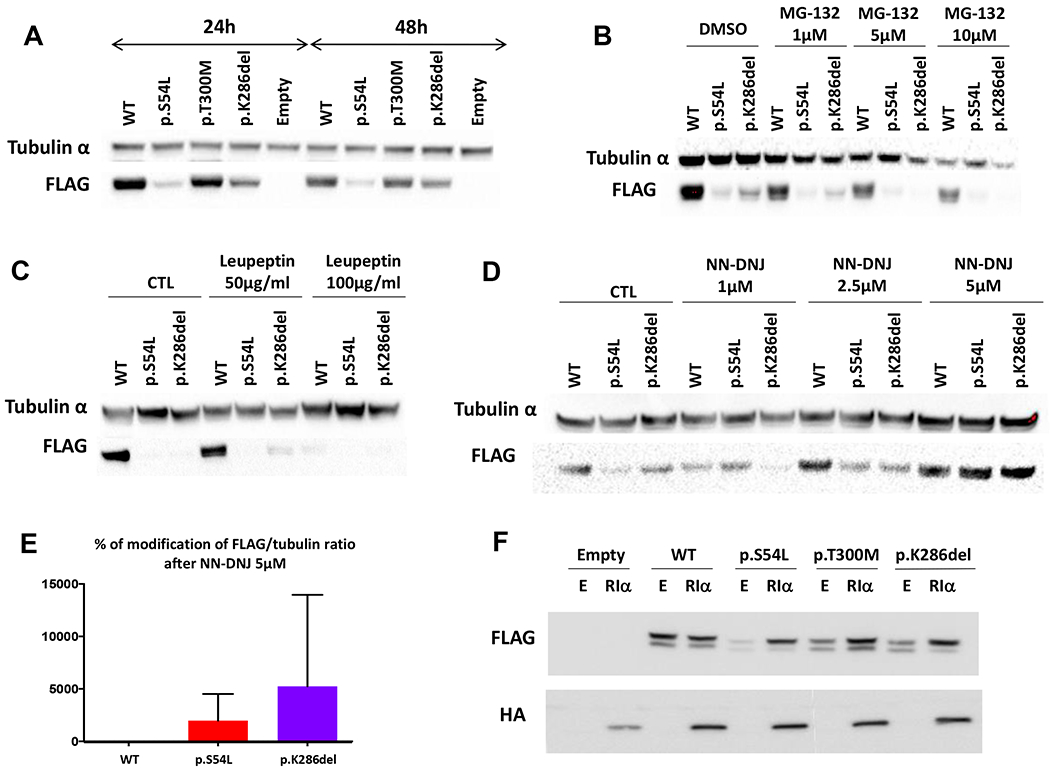
A. At 24 and 48 hours, in vitro expression of the p.S54L and p.K286del variants in HEK293 cells shows decreased protein presence compared to wild-type (WT). The p.T300M variant shows normal levels at both time points. B. Treatment of cells with proteasome inhibitor MG-132 did not increase the protein levels of the p.S54L and p.K286del variants. C. Likewise, treatment of cells with the lysosomal inhibitor leupeptin did not increase the protein levels of the p.S54L and p.K286del variants. D. Increasing amounts of the chaperone NN-DNJ molecule did stabilize the protein amounts of the p.S54L and p.K286del variants. E. Significant increase in the protein presence of the p.S54L and p.K286del variants after 5μM of the NN-DNJ (P < 0.05). F. Efficiency of co-transfection with RIα to re-establish the expression of the two mutants. E stands for Empty vector; Cβ vector was tagged with FLAG and RIα with HA; One representative experiment is shown (n=3).
PKA activity
We measured PKA activity both with co-expressed RIα (Figure 3A) or RIIβ (Figure 3B), the main PKA regulatory subunit binding to Cβ. The p.K286del variant in all experiments had the highest basal PKA activity (non-cAMP-stimulated), even higher than the previously reported p.S54L mutant (Espiard, et al. 2018). Accordingly, and unlike the p.S54L defect, the p.K286del failed to stimulate further upon exposure to cAMP. The stimulated activity of both mutants has to be interpreted with caution, since after stimulation by cAMP, the release from the regulatory subunit may lead to degradation of the mutant catalytic subunit, as previously shown for the p.S54L variant (Espiard, et al. 2018). Thus, the stimulated activity of the mutant protein may be underestimated. When co-expressed with RIα (Figure 3A) or RIIβ (Figure 3B), the p.T300M variant had PKA activity similar to that of the WT protein, both at baseline and after cAMP stimulation.
Figure 3. PKA activity in HEK293 cell lysates and immunohistochemistry in adrenal tissue from subject with p.T300M variant compared to controls.
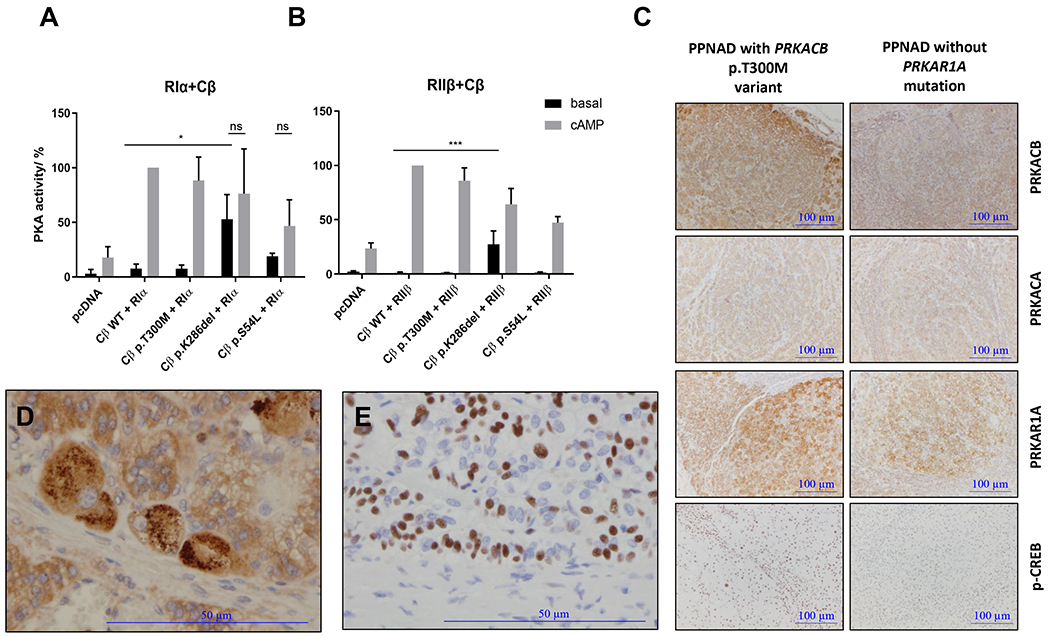
PKA activity was measured with co-expressed RIα (A) or RIIβ (B). The p.K286del variant had the highest baseline PKA activity, higher than p.S54L; unlike the p.S54L defect, the p.K286del failed to stimulate further upon exposure to cAMP. The p.T300M variant was not different from the baseline when co-expressed with RIα (A) or RIIβ (B). Data from 4 independent measurements are represented as mean ± SD and analyzed by two-way ANOVA with Tukey’s post hoc test for multiple comparisons, *P < 0.05 and ***P < 0.001 compared to basal Cβ wild-type (WT), ns stands for non-significant comparing the basal and the stimulated state (cAMP) of the same mutant. Representative Western Blots are indicated in Supplementary Figure 1. C. Adrenal tissue from the subject bearing the p.T300M variant expressed higher PRKACB protein with significant presence of phosphorylated CREB (p-CREB) compared to the adrenal gland from a PPNAD without PRKAR1A mutation. Magnification x40 of another immunostaining of PRKACB (D) and p-CREB presence (E) in the adrenal tissue of the subject bearing the p.T300M variant.
The immunohistochemistry of PRKACB, PRKACA and PRKAR1A in the adrenal tissue from the subject bearing the p.T300M variant showed a higher expression of the PRKACB protein (PRKACB, Figure 3C; magnification, Figure 3D) compared to PPNAD without PRKAR1A mutation. There was no PRKACB in the nucleus. A significant presence of phosphorylated CREB (p-CREB, Figure 3C; magnification, Figure 3E) was also observed in this subject compared to the adrenal gland from a subject without PRKAR1A mutation.
DISCUSSION
The Cβ catalytic subunit of PKA is expressed widely but at levels that are significantly lower than the Cα, the principal PKA catalytic subunit (Soberg, et al. 2013). Cβ was first identified in brain using Cα as a bait (Uhler, et al. 1986). When Prkaca was knocked-out in mice (Skalhegg, et al. 2002), Prkacab rescued the phenotype only in part and to varying degrees in different tissues (Huang, et al. 2002; Skalhegg, et al. 2002; Yin, et al. 2011); in the brain of Prkacb−/− mice, PKA activity was reduced by 26%, despite a compensatory increase in Cα (Howe, et al. 2002). Therefore, while global knockout of Cα is almost always lethal (Skalhegg, et al. 2002), deficiency of Cβ (which shares more than 90% sequence identity with Cα) leads only to minor phenotypes (Enns, et al. 2010; Enns, et al. 2009; Howe, et al. 2002; London, et al. 2019). These include lack of age‐related weight gain, resistance to diet-induced obesity, increased catecholaminergic activity, and protection against angiotensin II -induced cardiac hypertrophy and heart dysfunction (Enns, et al. 2010; Enns, et al. 2009; London, et al. 2019).
One challenge to studying Cβ function is that its contribution to total cellular PKA activity is much less than that of Ca. In addition, the PRKACB gene also undergoes extensive alternative splicing in a tissue-specific manner: there are the Cβ1, Cβ2, Cβ3 and Cβ4 isoforms, and the a, b, and c forms of Cβ3 and Cβ4 (Orstavik, et al. 2001). Only Cβ1 is ubiquitously expressed, and Cβ2 is present mainly in lymphocytes, while expression of Cβ3 and Cβ4 is limited to neuronal cells; tissue-specific deficiency of the respective Cβ isoform in mice leads to a cell-specific phenotype (Guthrie, et al. 1997; Moen, et al. 2017a). It appears that the two PKA catalytic subunits are associated with unique regulatory networks (Ihara, et al. 2017; Wu, et al. 2002) and PRKACB may have an independent role in skin pigmentation and various neoplasias, such as lymphoma, leukemia, thyroid and prostate cancer (Huang, et al. 2018; Moen, et al. 2017b; Saternus, et al. 2015; Wang, et al. 2020; Wu, et al. 2018).
In our present work, we screened a cohort of patients with possible PKA subunit defects and identified two subjects, one with what appears to be a new skeletal syndrome associated with developmental delay who carried the p.K286del PRKACB variant, and another with a PPNAD-like adrenal lesion and CS (Courcoutsakis, et al. 2004), who carried the p.T300M variant of the PRKACB protein (or p.T347M in another PRKACB isoform NM_182948.4). When the two new variants were tested along site the p.S54L one, we found that at least the p.K286del variant conferred PRKACB over-activity, whereas the data remain inconclusive for p.T300M. The p.K286del variant, albeit unstable when expressed in vitro (Figure 2), leads to high baseline PKA activity (Figure 3). In almost all of its investigated features, the p.K286del variant is more stable than p.S54L (Figure 2A) and appears to be stabilized by chaperone NN-DNJ (Figure 2E), and it has higher baseline PKA activity, whether it is co-expressed with RIα (Figure 3A) or RIIβ (Figure 3B). These data suggest a role for chaperone molecules in stabilizing defective PRKACB in cells.
The possible involvement of increased PRKACB expression and/or signaling in bone pathology has been suggested by animal studies (Liu, et al. 2016; Molyneux, et al. 2010; Tsang, et al. 2010): whenever Prkar1a and Prkaca were decreased, there was consistent upregulation of Prkacb, along with upregulation of the RII regulatory subunits (Liu, et al. 2016; Liu, et al. 2015; Tsang, et al. 2010). Decrease in either type of RII regulatory subunit mitigated the bone and other lesions in Prkar1a-deficient mice (Liu, et al. 2015), and treatment with celecoxib led to improvement in the bone phenotype that was also associated with decreased Prkacb expression (Saloustros, et al. 2017).
The functional data for p.T300M do not strongly support its pathogenicity: this variant had WT-like PKA activity when co-expressed with RIa or RIIβ. Despite the lack of more significant data for the p.T300M PRKACB variant, the subject’s adrenal tissue did show abundant expression of PRKACB and overrepresentation of p-CREB, when compared to other control tissues (Figure 3C). The absence of PRKACB nuclear localization suggests again the possible non-pathological role of this variant. At mRNA level, higher expression of RIa and Ca subunits was observed in the adrenal tissue of this subject when compared to conditions presenting PRKAR1A deletion or PRKACA duplication (see Supplementary Figure 5). The subject’s father was a carrier for the same p.T300M variant and he remains unaffected to date (although not biochemically screened); however, it is not unusual for male carriers of PKA signaling defects to not show adrenal disease, as we have shown in other settings (Bertherat, et al. 2009; Horvath, et al. 2010). The p.T300M variant is uncommon among the European and American populations but has a higher MAF among South Asians.
Thus, the role of PRKACB in adrenocortical development remains unclear, as the p.S54L variant was somatic (Espiard, et al. 2018). In terms of PRKACB’s effects on adrenocortical cell proliferation, however, the p.S54L data were strong and confirmed this variant’s high baseline PKA activity and associated increased proliferative capacity (Espiard, et al. 2018). In our experiments (Figure 3A and 3B), p.S54L showed indeed increased baseline PKA activity and relative unresponsiveness to cAMP, as is the case for activating PRKACA mutations (Beuschlein, et al. 2014; Di Dalmazi, et al. 2014; Stratakis 2014). Since the p.T300M studies do not show an increase in PKA activity, additional studies are needed to see if increased PRKACB expression in the adrenal cortex leads to hyperplasia as the one seen in our subject (Courcoutsakis, et al. 2004) and in carriers of germline PRKACA copy number gains (Carney, et al. 2015; Lodish, et al. 2015).
The facts that genomic PRKACB overexpression was seen along PRKACA defects in papillary neoplasms of the pancreas and bile duct (Singhi, et al. 2020) and that myxomas developed in the context of PRKACB copy number gain (Forlino, et al. 2014), just like the ones that formed due to somatic PRKACA defects (Stratakis 2018; Tseng, et al. 2017), suggest that PRKACB abnormalities are at least as capable of producing neoplasms as PRKACA defects are. The mechanism, however, remains unclear and maybe different than PRKACA: our data, for example, suggest that the p.T300M PRKACB may prefer RIIβ as a partner rather than RIα (Figure 3A and 3B). This tendency for a lower inhibition of RIIβ on p.T300M is concordant with our previous findings where RIIβ have a higher inhibition capacity on Cβ than RIα (Espiard, et al. 2018). The unique quaternary structures of the holoenzymes may explain the effect of some mutations on binding to one R subunit as previously described (Bathon, et al. 2019). PRKACB variants may also lead to a PKA enzyme that has novel molecular partners and thus, their effects maybe beyond those on enzymatic activity, as has been described for PRKACA (Bathon, et al. 2019; Stratakis 2019).
In conclusion, this investigation reveals two germline variants of the Cβ PKA catalytic subunit in two unrelated subjects, one with what appears to be a new syndrome of skeletal dysplasia, developmental delay, and other defects, and another with cortisol-producing micronodular adrenocortical hyperplasia. Although it remains unclear whether p.T300M is pathogenic, the data presented in this report along with our previously published studies on this gene, suggest that PRKACB should be included in the candidate genes for suspected PKA signaling defects in a variety of phenotypes that include adrenal or skeletal abnormalities, as well as possibly diseases of other systems where PKA signaling is involved.
Supplementary Material
Supplementary Figure 1. Expression of wild-type (WT) and mutant PRKACB proteins with co-expressed RIα or RIIβ. HEK293 cells were co-transfected with constant RIα (A) or RIIβ (B) and with WT or p.S54L, p.K286 del and p.T300M PRKACB vectors. Similar expression of mutant and WT PRKACB proteins in both (A) and (B).
Supplementary Figure 2. Confirmation of PRKACB p.K286del variant in subject #1. A. Pedigree and Sanger sequencing of subject’s #1 family showing the PRKACB p.K286del variant. B. NGS reads showing the PRKACB p.K286del variant.
Supplementary Figure 3. PRKACB missense p.T300M variant in subject #2. A. Sanger sequencing of subject’s #2 germline DNA showing the PRKACB p.T300M variant. B. NGS reads showing the PRKACB p.T300M variant in germline and somatic level.
Supplementary Figure 4. Somatic variants of PRKACB gene in different tissues. Data generated by the TGCA Research Network.
Supplementary Figure 5. Relative expression of PKA subunits in adrenal tissue of the subject harboring PRKACB p.T300M and other subjects with several defects. Samples were run in technical triplicates during qPCR and analyses done using ΔΔCT method. Values for subject harboring PRKACB p.T300M variant and subjects with PRKAR1A deletion or PRKACA duplication are expressed relative to control. GAPDH was used as a housekeeping gene.
Supplementary Table 1. Variants detected by sequencing in PRKACB gene in the cohort of 148 subjects.
Supplementary Table 2. Rare variants in conserved positions of subject #2
Supplementary Table 3. Multiple sequence alignment based in the PRKACB isoform NM_002731.3
Acknowledgments:
We thank Dr. Rhonda Schnur (Cooper University Hospital, Camden, NJ) and Dr. Emily Germain-Lee (University of Connecticut School of Medicine and Connecticut Children’s Medical Center, Farmington, Conn) for their contributions to the clinical characterization of subject #1. The results of the identified somatic variants of PRKACB gene in different tissues here are in whole or part based upon data generated by the TCGA Research Network: https://www.cancer.gov/tcga.
Funding:
This work was supported by the Intramural Research Program, Eunice Kennedy Shriver National Institute of Child Health & Human Development, National Institutes of Health (project number Z1A 29 HD008920)
Declaration of interest:
C.A.S. holds patent on the PRKAR1A, PDE11A and GPR101 genes and/or their function and his laboratory has received research funding from Pfizer Inc. F.R.F holds patent on the GPR101 gene and/or its function. The remaining authors have nothing to disclose.
REFERENCES
- Bathon K, Weigand I, Vanselow JT, Ronchi CL, Sbiera S, Schlosser A, Fassnacht M & Calebiro D 2019. Alterations in Protein Kinase A Substrate Specificity as a Potential Cause of Cushing Syndrome. Endocrinology 160 447–459. [DOI] [PubMed] [Google Scholar]
- Bertherat J, Groussin L, Sandrini F, Matyakhina L, Bei T, Stergiopoulos S, Papageorgiou T, Bourdeau I, Kirschner LS, Vincent-Dejean C, et al. 2003. Molecular and functional analysis of PRKAR1A and its locus (17q22-24) in sporadic adrenocortical tumors: 17q losses, somatic mutations, and protein kinase A expression and activity. Cancer Res 63 5308–5319. [PubMed] [Google Scholar]
- Bertherat J, Horvath A, Groussin L, Grabar S, Boikos S, Cazabat L, Libe R, Rene-Corail F, Stergiopoulos S, Bourdeau I, et al. 2009. Mutations in regulatory subunit type 1A of cyclic adenosine 5’-monophosphate-dependent protein kinase (PRKAR1A): phenotype analysis in 353 patients and 80 different genotypes. J Clin Endocrinol Metab 94 2085–2091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beuschlein F, Fassnacht M, Assie G, Calebiro D, Stratakis CA, Osswald A, Ronchi CL, Wieland T, Sbiera S, Faucz FR, et al. 2014. Constitutive activation of PKA catalytic subunit in adrenal Cushing’s syndrome. N Engl J Med 370 1019–1028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calebiro D, Bathon K & Weigand I 2017. Mechanisms of Aberrant PKA Activation by Calpha Subunit Mutations. Horm Metab Res 49 307–314. [DOI] [PubMed] [Google Scholar]
- Calebiro D, Hannawacker A, Lyga S, Bathon K, Zabel U, Ronchi C, Beuschlein F, Reincke M, Lorenz K, Allolio B, et al. 2014. PKA catalytic subunit mutations in adrenocortical Cushing’s adenoma impair association with the regulatory subunit. Nat Commun 5 5680. [DOI] [PubMed] [Google Scholar]
- Carney JA, Lyssikatos C, Lodish MB & Stratakis CA 2015. Germline PRKACA amplification leads to Cushing syndrome caused by 3 adrenocortical pathologic phenotypes. Hum Pathol 46 40–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carney JA, Boccon-Gibod L, Jarka DE, Tanaka Y, Swee RG, Unni KK & Stratakis CA 2001. Osteochondromyxoma of bone: a congenital tumor associated with lentigines and other unusual disorders. Am J Surg Pathol 25 164–176. [DOI] [PubMed] [Google Scholar]
- Courcoutsakis NA, Patronas NJ, Cassarino D, Griffin K, Keil M, Ross JL, Carney JA & Stratakis CA 2004. Hypodense nodularity on computed tomography: novel imaging and pathology of micronodular adrenocortical hyperplasia associated with myelolipomatous changes. J Clin Endocrinol Metab 89 3737–3738. [DOI] [PubMed] [Google Scholar]
- Di Dalmazi G, Kisker C, Calebiro D, Mannelli M, Canu L, Arnaldi G, Quinkler M, Rayes N, Tabarin A, Laure Jullie M, et al. 2014. Novel somatic mutations in the catalytic subunit of the protein kinase A as a cause of adrenal Cushing’s syndrome: a European multicentric study. J Clin Endocrinol Metab 99 E2093–2100. [DOI] [PubMed] [Google Scholar]
- Enns LC, Bible KL, Emond MJ & Ladiges WC 2010. Mice lacking the Cbeta subunit of PKA are resistant to angiotensin II-induced cardiac hypertrophy and dysfunction. BMC Res Notes 3 307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Enns LC, Morton JF, Mangalindan RS, McKnight GS, Schwartz MW, Kaeberlein MR, Kennedy BK, Rabinovitch PS & Ladiges WC 2009. Attenuation of age-related metabolic dysfunction in mice with a targeted disruption of the Cbeta subunit of protein kinase A. J Gerontol A Biol Sci Med Sci 64 1221–1231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Espiard S, Knape MJ, Bathon K, Assie G, Rizk-Rabin M, Faillot S, Luscap-Rondof W, Abid D, Guignat L, Calebiro D, et al. 2018. Activating PRKACB somatic mutation in cortisol-producing adenomas. JCI Insight 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forlino A, Vetro A, Garavelli L, Ciccone R, London E, Stratakis CA & Zuffardi O 2014. PRKACB and Carney complex. N Engl J Med 370 1065–1067. [DOI] [PubMed] [Google Scholar]
- Guthrie CR, Skalhegg BS & McKnight GS 1997. Two novel brain-specific splice variants of the murine Cbeta gene of cAMP-dependent protein kinase. J Biol Chem 272 29560–29565. [DOI] [PubMed] [Google Scholar]
- Honeyman JN, Simon EP, Robine N, Chiaroni-Clarke R, Darcy DG, Lim II, Gleason CE, Murphy JM, Rosenberg BR, Teegan L, et al. 2014. Detection of a recurrent DNAJB1-PRKACA chimeric transcript in fibrolamellar hepatocellular carcinoma. Science 343 1010–1014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horvath A, Bertherat J, Groussin L, Guillaud-Bataille M, Tsang K, Cazabat L, Libe R, Remmers E, Rene-Corail F, Faucz FR, et al. 2010. Mutations and polymorphisms in the gene encoding regulatory subunit type 1-alpha of protein kinase A (PRKAR1A): an update. Hum Mutat 31 369–379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howe DG, Wiley JC & McKnight GS 2002. Molecular and behavioral effects of a null mutation in all PKA C beta isoforms. Mol Cell Neurosci 20 515–524. [DOI] [PubMed] [Google Scholar]
- Huang Y, Roelink H & McKnight GS 2002. Protein kinase A deficiency causes axially localized neural tube defects in mice. J Biol Chem 277 19889–19896. [DOI] [PubMed] [Google Scholar]
- Huang Y, Huang Y, Zhang L, Chang A, Zhao P, Chai X & Wang J 2018. Identification of crucial genes and prediction of small molecules for multidrug resistance of Hodgkin’s lymphomas. Cancer Biomark 23 495–503. [DOI] [PubMed] [Google Scholar]
- Ihara T, Hosokawa Y, Kumazawa K, Ishikawa K, Fujimoto J, Yamamoto M, Muramkami T, Goshima N, Ito E, Watanabe S, et al. 2017. An in vivo screening system to identify tumorigenic genes. Oncogene 36 2023–2029. [DOI] [PubMed] [Google Scholar]
- Kirschner LS, Carney JA, Pack SD, Taymans SE, Giatzakis C, Cho YS, Cho-Chung YS & Stratakis CA 2000. Mutations of the gene encoding the protein kinase A type I-alpha regulatory subunit in patients with the Carney complex. Nat Genet 26 89–92. [DOI] [PubMed] [Google Scholar]
- Linglart A, Menguy C, Couvineau A, Auzan C, Gunes Y, Cancel M, Motte E, Pinto G, Chanson P, Bougneres P, et al. 2011. Recurrent PRKAR1A mutation in acrodysostosis with hormone resistance. N Engl J Med 364 2218–2226. [DOI] [PubMed] [Google Scholar]
- Liu S, Shapiro JM, Saloustros E & Stratakis CA 2016. Bone Abnormalities in Mice with Protein Kinase A (PKA) Defects Reveal a Role of Cyclic AMP Signaling in Bone Stromal Cell-Dependent Tumor Development. Horm Metab Res 48 714–725. [DOI] [PubMed] [Google Scholar]
- Liu S, Saloustros E, Mertz EL, Tsang K, Starost MF, Salpea P, Faucz FR, Szarek E, Nesterova M, Leikin S, et al. 2015. Haploinsufficiency for either one of the type-II regulatory subunits of protein kinase A improves the bone phenotype of Prkar1a+/− mice. Hum Mol Genet 24 6080–6092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lodish MB, Yuan B, Levy I, Braunstein GD, Lyssikatos C, Salpea P, Szarek E, Karageorgiadis AS, Belyavskaya E, Raygada M, et al. 2015. Germline PRKACA amplification causes variable phenotypes that may depend on the extent of the genomic defect: molecular mechanisms and clinical presentations. Eur J Endocrinol 172 803–811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- London E, Noguchi A, Springer D, Faidas M, Gavrilova O, Eisenhofer G & Stratakis CA 2019. The Catalytic Subunit beta of PKA Affects Energy Balance and Catecholaminergic Activity. J Endocr Soc 3 1062–1078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moen LV, Sener Z, Volchenkov R, Svarstad AC, Eriksen AM, Holen HL & Skalhegg BS 2017a. Ablation of the Cbeta2 subunit of PKA in immune cells leads to increased susceptibility to systemic inflammation in mice. Eur J Immunol 47 1880–1889. [DOI] [PubMed] [Google Scholar]
- Moen LV, Ramberg H, Zhao S, Grytli HH, Sveen A, Berge V, Skotheim RI, Tasken KA & Skalhegg BS 2017b. Observed correlation between the expression levels of catalytic subunit, Cbeta2, of cyclic adenosine monophosphate-dependent protein kinase and prostate cancer aggressiveness. Urol Oncol 35 111.e111–111.e118. [DOI] [PubMed] [Google Scholar]
- Molyneux SD, Di Grappa MA, Beristain AG, McKee TD, Wai DH, Paderova J, Kashyap M, Hu P, Maiuri T, Narala SR, et al. 2010. Prkar1a is an osteosarcoma tumor suppressor that defines a molecular subclass in mice. J Clin Invest 120 3310–3325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orstavik S, Reinton N, Frengen E, Langeland BT, Jahnsen T & Skalhegg BS 2001. Identification of novel splice variants of the human catalytic subunit Cbeta of cAMP-dependent protein kinase. Eur J Biochem 268 5066–5073. [DOI] [PubMed] [Google Scholar]
- Patronas Y, Horvath A, Greene E, Tsang K, Bimpaki E, Haran M, Nesterova M & Stratakis CA 2012. In vitro studies of novel PRKAR1A mutants that extend the predicted RIalpha protein sequence into the 3’-untranslated open reading frame: proteasomal degradation leads to RIalpha haploinsufficiency and Carney complex. J Clin Endocrinol Metab 97 E496–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saloustros E, Liu S, Mertz EL, Bhattacharyya N, Starost MF, Salpea P, Nesterova M, Collins M, Leikin S & Stratakis CA 2017. Celecoxib treatment of fibrous dysplasia (FD) in a human FD cell line and FD-like lesions in mice with protein kinase A (PKA) defects. Mol Cell Endocrinol 439 165–174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salpea P, Horvath A, London E, Faucz FR, Vetro A, Levy I, Gourgari E, Dauber A, Holm IA, Morrison PJ, et al. 2014. Deletions of the PRKAR1A locus at 17q24.2-q24.3 in Carney complex: genotype-phenotype correlations and implications for genetic testing. J Clin Endocrinol Metab 99 E183–188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saternus R, Pilz S, Graber S, Kleber M, Marz W, Vogt T & Reichrath J 2015. A closer look at evolution: Variants (SNPs) of genes involved in skin pigmentation, including EXOC2, TYR, TYRP1, and DCT, are associated with 25(OH)D serum concentration. Endocrinology 156 39–47. [DOI] [PubMed] [Google Scholar]
- Singhi AD, Wood LD, Parks E, Torbenson MS, Felsenstein M, Hruban RH, Nikiforova MN, Wald AI, Kaya C, Nikiforov YE, et al. 2020. Recurrent Rearrangements in PRKACA and PRKACB in Intraductal Oncocytic Papillary Neoplasms of the Pancreas and Bile Duct. Gastroenterology 158 573–582.e572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skalhegg BS, Huang Y, Su T, Idzerda RL, McKnight GS & Burton KA 2002. Mutation of the Calpha subunit of PKA leads to growth retardation and sperm dysfunction. Mol Endocrinol 16 630–639. [DOI] [PubMed] [Google Scholar]
- Soberg K, Jahnsen T, Rognes T, Skalhegg BS & Laerdahl JK 2013. Evolutionary paths of the cAMP-dependent protein kinase (PKA) catalytic subunits. PLoS One 8 e60935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stratakis CA 2014. E pluribus unum? The main protein kinase A catalytic subunit (PRKACA), a likely oncogene, and cortisol-producing tumors. J Clin Endocrinol Metab 99 3629–3633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stratakis CA 2018. Cyclic AMP-dependent protein kinase catalytic subunit A (PRKACA): the expected, the unexpected, and what might be next. J Pathol 244 257–259. [DOI] [PubMed] [Google Scholar]
- Stratakis CA 2019. Called and Uncalled for Functions of the Main Catalytic Subunit of Protein Kinase A: One Enzyme, Many Faces. Endocrinology 160 1674–1676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tasken K & Aandahl EM 2004. Localized effects of cAMP mediated by distinct routes of protein kinase A. Physiol Rev 84 137–167. [DOI] [PubMed] [Google Scholar]
- Taylor SS, Ilouz R, Zhang P & Kornev AP 2012. Assembly of allosteric macromolecular switches: lessons from PKA. Nat Rev Mol Cell Biol 13 646–658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsang KM, Starost MF, Nesterova M, Boikos SA, Watkins T, Almeida MQ, Harran M, Li A, Collins MT, Cheadle C, et al. 2010. Alternate protein kinase A activity identifies a unique population of stromal cells in adult bone. Proc Natl Acad Sci U S A 107 8683–8688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tseng IC, Huang WJ, Jhuang YL, Chang YY, Hsu HP & Jeng YM 2017. Microinsertions in PRKACA cause activation of the protein kinase A pathway in cardiac myxoma. J Pathol 242 134–139. [DOI] [PubMed] [Google Scholar]
- Turan S & Bastepe M 2015. GNAS Spectrum of Disorders. Curr Osteoporos Rep 13 146–158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uhler MD, Chrivia JC & McKnight GS 1986. Evidence for a second isoform of the catalytic subunit of cAMP-dependent protein kinase. J Biol Chem 261 15360–15363. [PubMed] [Google Scholar]
- Wang Y, Wang B, Zhou H, Zhang X, Qian X & Cui J 2020. MicroRNA-384 Inhibits the Progression of Papillary Thyroid Cancer by Targeting PRKACB. Biomed Res Int 2020 4983420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu DM, Wen X, Han XR, Wang S, Wang YJ, Shen M, Fan SH, Zhang ZF, Shan Q, Li MQ, et al. 2018. Role of Circular RNA DLEU2 in Human Acute Myeloid Leukemia. Mol Cell Biol 38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu KJ, Mattioli M, Morse HC 3rd & Dalla-Favera 2002. c-MYC activates protein kinase A (PKA) by direct transcriptional activation of the PKA catalytic subunit beta (PKA-Cbeta) gene. Oncogene 21 7872–7882. [DOI] [PubMed] [Google Scholar]
- Yin Z, Pringle DR, Jones GN, Kelly KM & Kirschner LS 2011. Differential role of PKA catalytic subunits in mediating phenotypes caused by knockout of the Carney complex gene Prkar1a. Mol Endocrinol 25 1786–1793. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure 1. Expression of wild-type (WT) and mutant PRKACB proteins with co-expressed RIα or RIIβ. HEK293 cells were co-transfected with constant RIα (A) or RIIβ (B) and with WT or p.S54L, p.K286 del and p.T300M PRKACB vectors. Similar expression of mutant and WT PRKACB proteins in both (A) and (B).
Supplementary Figure 2. Confirmation of PRKACB p.K286del variant in subject #1. A. Pedigree and Sanger sequencing of subject’s #1 family showing the PRKACB p.K286del variant. B. NGS reads showing the PRKACB p.K286del variant.
Supplementary Figure 3. PRKACB missense p.T300M variant in subject #2. A. Sanger sequencing of subject’s #2 germline DNA showing the PRKACB p.T300M variant. B. NGS reads showing the PRKACB p.T300M variant in germline and somatic level.
Supplementary Figure 4. Somatic variants of PRKACB gene in different tissues. Data generated by the TGCA Research Network.
Supplementary Figure 5. Relative expression of PKA subunits in adrenal tissue of the subject harboring PRKACB p.T300M and other subjects with several defects. Samples were run in technical triplicates during qPCR and analyses done using ΔΔCT method. Values for subject harboring PRKACB p.T300M variant and subjects with PRKAR1A deletion or PRKACA duplication are expressed relative to control. GAPDH was used as a housekeeping gene.
Supplementary Table 1. Variants detected by sequencing in PRKACB gene in the cohort of 148 subjects.
Supplementary Table 2. Rare variants in conserved positions of subject #2
Supplementary Table 3. Multiple sequence alignment based in the PRKACB isoform NM_002731.3