

Abstract
It is well demonstrated that activation of renal (pro)renin receptor (PRR) contributes to angiotensin II (AngII)-induced hypertension. Relatively, less is known for the function of soluble PRR (sPRR), the extracellular domain of PRR, primarily generated by site-1 protease (S1P) and furin. Moreover, the relationship between PRR/sPRR and the renin-angiotensin system (RAS) has been debated. In the present study, we employed CRISPR/Cas9 strategy to generate mice with mutegenesis of the overlapping cleavage site for both proteases in PRR (termed as PRRR279V/L282V) to examine the phenotype during AngII infusion with particular emphasis on circulating and intrarenal renin status. PRRR279V/L282V mice exhibited a reduction of sPRR level in plasma by ~53% and in the kidney by ~82%, were fertile and had no gross developmental abnormalities. At basal condition, PRRR279V/L282V mice had drastically suppressed renin levels from plasma, urine, and the kidney as compared with WT controls. The hypertensive response of PRRR279V/L282V to AngII infusion was blunted in parallel with attenuated response of intrarenal renin and renal medullary α-ENaC expression. By using Ussing chamber technique, primary collecting duct (CD) cells from PRRR279V/L282V mice exhibited blunted response of ENaC activity to AngII as compared with WT cells. Together, these results represent strong evidence favoring sPRR as a mediator of AngII-induced hypertension and a master regulator of renin expression. Therefore, PRR should be considered as an integrative member of the RAS.
Keywords: Soluble (pro)renin receptor, site-1 protease, angiotensin II, renin activity, epithelial sodium channel
Summary
We for the first time report the generation and characterization of a novel mouse model with mutagenesis of the cleavage site of PRR during AngII-induced hypertension. The results demonstrated an essential role of sPRR in mediating the hypertensive response to AngII infusion in mice. Moreover, we provided a mechanism of this phenomenon, which involves activation of intrarenal RAS and selective stimulation of renal medullary α-ENaC expression. Besides the control of intrarenal RAS, sPRR also serves as a key regulator of systemic renin level. Overall, the present study provides strong evidence supporting the intrinsic relationship between PRR/sPRR and the RAS.
Introduction
(Pro)renin receptor (PRR) was originally cloned as a specific transmembrane receptor for prorenin and renin implicated in regulation of tissue renin-angiotensin system (RAS) 1. Besides its association with RAS, PRR plays a complex role in that the cytoplasmic domain is an accessory protein (M8–9), a subunit of the vacuolar-type H+-ATPase (V-ATPase) (also designated ATP6ap2) 3 and it activates multiple signaling transduction pathways including MAP kinase and β-catenin signaling 12–14. More complex is that the extracellular domain of PRR is cleaved by proteases to generate a biological active soluble PRR (sPRR) 4, 15. Through multiple mechanisms, PRR participates in a wide variety of developmental and physiological processes involving multiple organs 14,16,17.
Within the kidney, PRR is abundantly expressed in intercalated cells of the collecting duct (CD) albeit with relatively lower expression in multiple nephron structures 8. Majority of studies show that deletion of PRR in renal tubules 9 or the CD 10, 11 impairs urine concentrating capability and attenuates hypertension development during AngII infusion. The prohypertensive role of PRR is dependent on activation of intrarenal renin-angiotensin system (RAS) and enhancement of α-ENaC expression 18–20. However, the detailed mechanism of how PRR is involved in BP regulation remains elusive. Emerging evidence reveals biological functions of sPRR in renal handling of Na+ and water. In this regard, exogenous administration of histidine-tagged sPRR (sPRR-His) directly upregulated expression of aquaporin-2 (AQP2) and α-ENaC both via activation of β-catenin signaling, contributing to enhancement of urine concentrating capability 22. Subsequently, the functional role of endogenous sPRR has been suggested by the use of a S1P inhibitor PF-429242 and the generation of renal tubule-wide S1P deletion 22. Although these experimental approaches are helpful in defining the function of sPRR, S1P cleaves other substrates than PRR 23. To gain more definitive evidence for the function of endogenous sPRR, we employed CRISP/Cas9 strategy to generate a novel mouse model of mutagenesis of the cleavage site of PRR and characterize the phenotype during AngII-induced hypertension.
Methods
The data that support the findings of this study are available from the corresponding author upon reasonable request.
Animal care.
All animals were cage-housed and maintained in a temperature-controlled room with a 12:12-hour light-dark cycle, with free access to tap water and standard rat chow. Animals were randomized into different experimental groups. All animal studies conducted in the present study were approved by the Animal Care and Use Committee at the University of Utah.
Generation of PRRR279V/L282V mice.
The mutant mice were generated by using CRISPR/Cas9-mediated genome engineering thought the service from Cygene. PRRR279V/R282V means that the amino acid arginine at the 279 position and the amino acid leucine at the 282 position of PRR were both mutated to valine. The mouse PRR gene contains 9 exons, with the ATG start codon in exon 1 and the TGA stop codon in exon 9. The codons for furin recognizing site: RKSR, and S1P recognizing site: RTIL are transcribed from the exon 8. To mutate these recognizing sites in PRR protein, donor oligos, guidance RNA (gRNA) and Cas9 mRNA were co-injected into fertilized eggs to generate PRRR279V & L282V knock-in C57Bl6 offsprings. The sequence of the donor oligos is: 5’-TACAGTCTCTATGGTGGGAACGCAGTGGTAGAGTTAGTGACTGTCAAATCATTCGACACATCTCTTGTGAGGAAGTCAGTGACCATCGTTGAAGCAAAACAAGAGGTGAGTAACTTTTTGGATCCTGCTTTAATGCATGAAGTTAGCTTACTT-3’. It is a homologous sequence with a targeting sequence, flanked by 153 bp combined on both sides. The targeting sequence: 5’-TCTCTTGTGAGGAAGTCAGTGACCATCGTTGAA-3’, was used to replace genomic sequence, 5’-TCCCTTGTGAGGAAGTCAAGGACCATCCTTGAG-3’, in the exon 8 of PRR gene by homology-directed repair. After this replacement, the amino acids 276RKSRTILE283 in PRR protein will be changed to 276RKSVTIVE283. The gRNA sequence matches forward strand of PRR gene: 5’-GAAGTCAAGGACCATCCTTGAGG-3’. F0 founder animals were identified by PCR followed by sequence analysis, which were bred to C57Bl6 mice to test germline transmission and F1 animal generation. Because of the infertility of the homozygous male, the heterozygous females were mated to wild type males (gifted from Transgenic Gene Targeting Facility, University of Utah, C57Bl6/CBA) to produce wild type and homozygous male alleles, as well as heterozygous females. The genotype of the alleles was identified by PCR followed by AvaII and AgsI restriction. The primers used for PCR were as follows: forward primer 5’-GTGAATACCAAGGGACAGTTTTAC-3’ and reverse primer 5’-CAGATTGCCAGGCATACAGCCAG-3’. The PCR was perform at 94°C 30sec, 55°C 30 sec, and 72°C 60 sec for 40 cycles. The expected size of PCR product is 383 bp. AvaII only cut the PCR product from wild type allele into two fragments of 163 bp and 220 bp. In contrast, AgsI only cut the PCR product from mutated allel into two fragments of 174 bp and 209 bp. The 383 bp PCR products from the two genotypes were further analyzed by DNA sequencing.
Mouse experiments.
Male 16 to 20-week-old PRRR279V/L282V mice and their littermate wild-type (WT) controls were studied. Five days prior to AngII treatment, under anesthesia by 2% isoflurane, the radiotelemetric device was implanted via catheterization of carotid artery. After the baseline blood pressure (BP) parameters were recorded, all mice were infused for 14 days with vehicle or AngII at 300 ng·kg−1·min−1 via a subcutaneously implanted mini-pump (Alzet model 1002, Alza). Mice were not disturbed during the BP recording period. BP was recorded for 4 h per day from 5:00PM to 9:00PM. On day 11 treatment of AngII, the mice were placed at metabolic cage for urine collection over 4 days. At the end of the experiment, 24-h urine was collected, and then under general anesthesia, blood was withdrawn by puncturing vena cava, and the kidney was harvested and cut into cortex and the inner medulla and snap frozen.
Renin enzyme activity assay.
Renin activity assay was performed as previously described 19. Briefly, renin activity in urine and renal inner medulla was determined by the delta value of the Ang I generation using an ELISA kit from the sample incubating at 4°C and 37°C for 1 h, respectively. Total renin content was measured with excessive angiotensinogen (AGT) plus trypsinization, and active renin content with excessive AGT. Urine and tissue samples were spiked with 1 M synthetic renin substrate tetradecapeptide (RST, R8129; Sigma-Aldrich, St. Louis, MO). After incubation at 37°C for 18 h, AngI generation was assayed by using an AngI EIA kit (S-1188; Peninsula Laboratories International, SanCarlos, CA), according to the manufacturer’s instructions. The values were expressed as nanograms per milliliter per hour of generated AngI. For measurement of total renin content, trypsinization was performed to activate prorenin to renin 24. The samples were incubated with trypsin derived from bovine pancreas (100 g/l, T1426, Sigma-Aldrich) in 37°C for 18 h. The reaction was then terminated with soybean trypsin inhibitor (100 g/l, T6522; Sigma-Aldrich) at 37°C for 1 h.
Immunobloting.
Renal tissues including the cortex and the inner medulla were lysed and subsequently sonicated in PBS that contained 1% Triton x-100, 250 μM phenylmethanesulfonyl fluoride (PMSF), 2 mM EDTA, and 5 mM dithiothrietol (DTT) (pH 7.5). Protein concentrations were determined by using Coomassie reagent. Forty μg of proteins for each sample was denatured in boiling water for 10 min, then separated by SDS-PAGE gel, and transferred onto nitrocellulose membranes. The blots were blocked overnight with 5% nonfat dry milk in Tris-buffered saline (TBS), followed by incubation for overnight with primary antibody. After being washed with TBS, blots were incubated with horseradish peroxidase (HRP)-conjugated secondary antibody and visualized using Enhanced Chemiluminescence (ECL). The blots were quantitated by using Imagepro-plus. Primary antibodies are as follows: rabbit anti-PRR antibody (cat no. HPA003156; Sigma), rabbit anti-α-ENaC antibody (cat no. SPC-403D; Stressmarq Biosciences), mouse anti-renin antibody (cat no. sc133145; Santa Cruz), rabbit anti-Na+-Cl− cotransporter (NCC) antibody (cat no. SPC-402D), rabbit anti-Na+-K+−2Cl−cotransporter (NKCC2) antibody (cat no. SPC-401), rabbit anti-sodium-hydrogen antiporter3 (NHE3) (cat no. SPC-400), mouse anti-β-actin antibody (cat no. A1978, Sigma).
qRT-PCR.
Total RNA isolation and reverse transcription were performed as previously described 19. Oligonucleotides were designed using Primer3 software (available at http://bioinfo.ut.ee/primer3-0.4.0/). Primers were as follows: for α-ENaC: 5’-gcgacaacaatccccaag-3’ (sense) and 5’-tgaagcgacaggtgaagatg-3’ (antisense); for β-ENaC: 5’-aagcacctgtaatgcccaag-3’ (sense) and 5’-atagcccatccccaccag-3’ (antisense); for γ-ENaC: 5’-cgaagaaactggtgggattt-3’ (sense) and 5’-gatggtggaaaagcgtgaag-3’ (antisense); for Renin: 5’-gatcaccatgaagggggtctctgt-3’ (sense) and 5’-gttcctgaagggattcttttgcac-3’ (antisense) and for GAPDH: 5’-gtcttcactaccatggagaagg-3’ (sense) and 5’-tcatggatgaccttggccag-3’ (antisense).
Primary cultures of mouse inner medullary collecting duct (IMCD) cells.
IMCD cells were prepared from 4-week-old mutant mice and their littermate WT mice as previously described19. The cells were grown in Transwells (catalog no. 29442–074; VWR International) with DMEM/F-12 medium containing 10% FBS, 0.5 μM 8-Br-cAMP, 130 mM NaCl, and 80 mM urea. Upon confluence, cells were serum-starved for 12 h, and then treated with AngII (1 μM) for 24 hours. After treatment, the cells were subjected to electrophysiolocial measurement of ENaC activity.
Electrophysiological measurements of transepithelial Na+ transport.
Electrophysiology experiments were performed on primary cultures of mouse IMCD cells after the cell monolayers reached confluence. Transepithelial Na+ transport was recorded by using the Ussing chamber device (Physiologic Instruments, San Diego, CA) as previously described 25, 26. Briefly, a Snapwell insert containing IMCD cells was mounted in the Ussing chamber. Both hemichambers were filled with a Krebs’ buffer solution containing 120 mM NaCl, 25 mM NaHCO3, 3.3 mM KH2PO4, 0.8 mM K2HPO4, 0.5 mM MgCl2, 10 mM HEPES, and 10 mM glucose (pH 7.4). The solution was continuously bubbled with 5% CO2/95% O2 gas mixture at 37 °C. In performing Ussing chamber technique, the voltage was clamped at zero using a VCC600 voltage-clamp apparatus (Physiologic Instruments), and then the short-circuit current (Isc) was recorded using Ag-AgCl electrode in agar brides. Positive Isc reflects the active transport of cation (Na+) from apical side to basolateral side of media or transport of anion (Cl−) from basolateral to apical side of media. After collecting the Isc values, 100 μM amiloride was added to all groups. Amiloride-sensitive component was taken as an index of ENaC activity.
Enzyme immunoassay.
The sPRR and prorenin/renin in biologic fluids were determined by using the following commercially available enzyme immunoassay kits according to the manufacturer’s instructions: the kits for sPRR (catalog no. JP27782; IBL, Toronto, ON, Canada), and prorenin/renin (Molecular Innovations, Novi, MI).
Statistics.
GraphPad Prism software (version 8.4) was used for data analysis Data is summarized as means ± SEM. All data points represent animals that were included in the statistical analyses. Sample sizes were determined on the basis of similar previous studies or pilot experiments. For BP experiment, the statistical significance was determined by using two-way ANOVA with repeated measurements. The other animal and cell culture experiments were performed by using regular two-way ANOVA with the Bonferroni test for multiple comparisons or by using unpaired two-tailed Student’s t test for 2 comparisons. A value of P < 0.05 was considered statistically significant.
Results
Creation and genotyping of mice with mutagenesis of the cleavage site of PRR
The physiological function of sPRR, the extracellular domain of PRR cleaved by S1P, has been suggested by pharmacological studies22, 27, 28. However, definitive evidence for this notion is still lacking. The present study attempted to employ CRISPR/Cas9 strategy to mutagenize the overlapping recognition site for S1P and furin in PRR to examine its impact on AngII-induced hypertension. The genotyping of PRRR279V/L282V mice was first analyzed by restriction enzyme digestion. The mutation resulted in digestion of the PCR product by Ags I instead of Ava II as seen in WT control (Fig. 1A). Following this, DNA sequencing confirmed the expected mutations in PRR gene (Fig. 1B). By immunostaining, PRRR279V/L282V mice had significantly decreased sPRR protein abundance in kidney (Fig. 1C), adipose tissue and adrenal glands (Fig. S1) accompanied by slightly increased full-length PRR abundance as compared with littermate WT controls. By ELISA, plasma sPRR was reduced by 53% in PRRR279V/L282V mice (Fig. 1D). PRRR279V/L282V mice was born at the expected Mendelian ratio without noticeable developmental abnormalities. The structural integrity of the mutant kidney appeared to be well maintained as evidenced by normal histology by Hematoxylin and Eosin (H&E) staining, and the lack of indices of autophagosome accumulation (Fig. S2). On a normal chow diet, PRRR279V/L282V mice developed obesity accompanied with elevated fasting blood glucose. The metabolic phenotype was pursued by a separate study (manuscript in preparation).
Figure. 1.
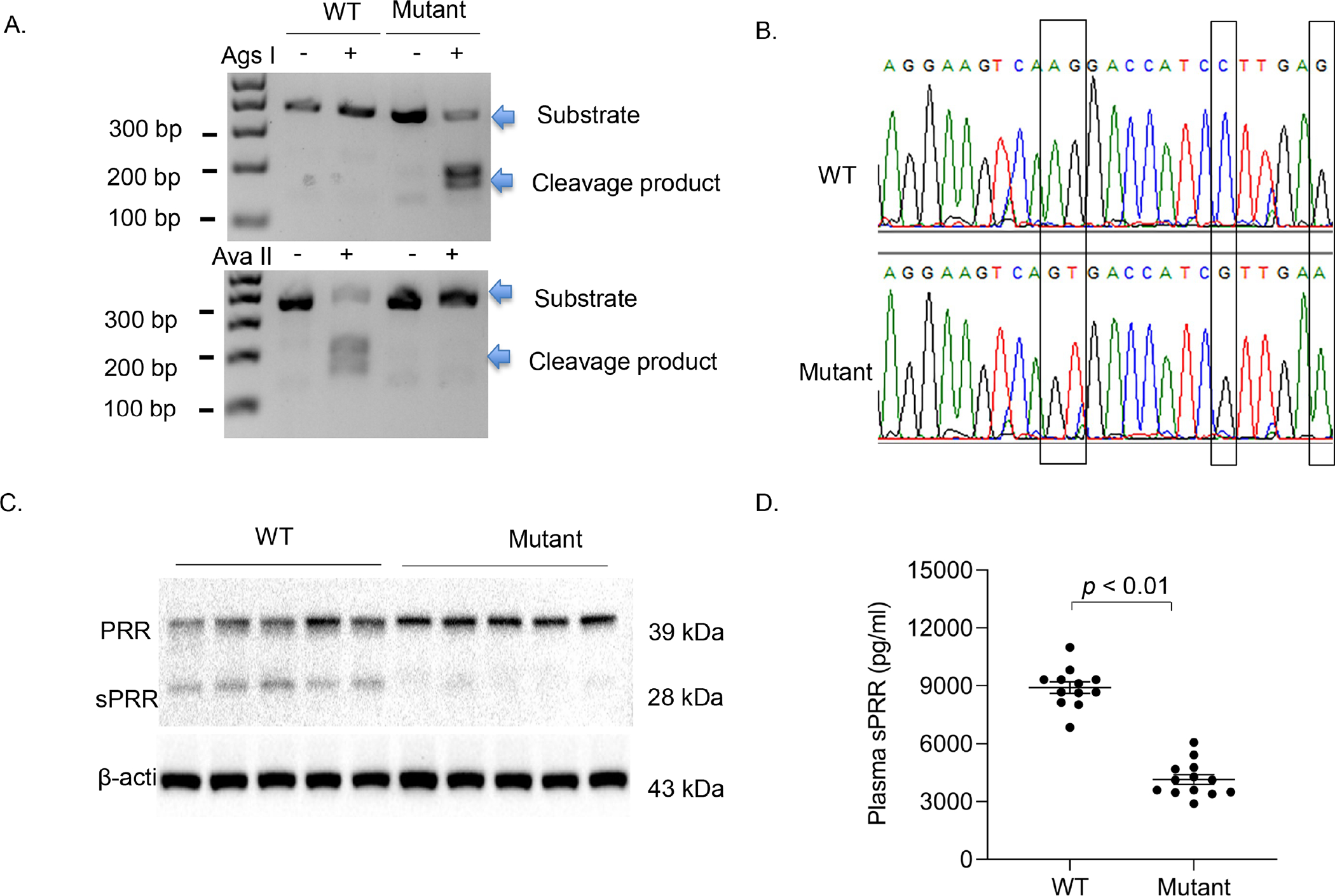
Genotyping of PRRR279V/L282V mice. Genotyping was first performed by PCR using tail DNA. A PCR product of 383 bp spanning the cleavage site was subjected to restriction enzyme digestion followed by DNA electrophoresis (A). The PCR product containing the mutant PRR allele became sensitive to Ags I but not Ava II whereas the product harboring the WT PRR allele remained sensitive to Ava II but not Ags I. (B) DNA sequencing analysis of the PCR products from WT and PRRR279V/L282V mice. (C) Immunoblotting analysis of renal PRR/sPRR expression in the two genotypes (n = 5 per each group). (D) ELISA analysis of plasma sPRR concentrations in the two genotypes (n = 12 per each group). The statistical significance was determined by using unpaired two-tailed Student’s t test for 2 comparisons, and the p values were indicated in the figure. Data are mean ± SE. WT, wild-type; Mutant, PRRR279V/L282V.
The BP response to AngII
Renal expression of PRR and production of sPRR are elevated by AngII in vivo and in vitro and renal medullary inhibition of PRR by PRR decoy inhibitor PRO20 or conditional PRR deletion in the kidney or the CD attenuated AngII-induced hypertension. Therefore, we examined the BP phenotype of PRRR279V/L282V mice during 14-day AngII infusion. Radiotelemetry was used to monitor daily changes in mean arterial pressure (MAP), systolic blood pressure (SBP), diastolic blood pressure (DBP), and heart rate (HR) during the first 10 days. AngII infusion at a pressor dose of 300 ng/kg/min gradually and significantly elevated MAP, SBP, DBP, and decreased HR in WT mice (day 10: delta MAP 27.9 ± 3.1 mmHg; delta SBP 26.9 ± 4.4 mmHg; delta DBP 25.1 ± 2.0 mmHg; delta HR −42.4 ± 6.4 beats/min). In contrast, the increases in BP parameters in Mutant/AngII group were less (day 10: delta MAP 13.4 ± 7.2 mmHg; delta SBP 11.5 ± 8.2 mmHg; delta DBP 11.3 ± 6.4 mmHg; delta HR −25.2 ± 10.4 beats/min). These results suggest that PRRR279V/L282V mice exhibited blunted hypertensive response to AngII infusion.
To address the potential vascular mechanism involved in the BP phenotype of PRRR279V/L282V mice, we examined the pressor response to acute AngII treatment under conscious condition using radiotelemetry. The s.c. injection of vehicle or AngII at 30 μg/kg within minutes induced a similar stress response in both WT and PRRR279V/L282V mice. Following the stress response, a second rise of BP was only observed in the AngII group but not the vehicle group, confirming the pressor effect induced by AngII injection. However, AngII-induced pressor response was comparable between the two genotypes (Fig. S3).
Twenty four-hour urine was collected on the last day of AngII treatment. Urine volume and urinary Na+ excretion was elevated in WT/AngII mice. A similar increase in urine volume was observed in PRRR279V/L282V/AngII mice. However, there was only a trend of increase in urinary Na+ excretion in the null mice that didn’t reach a statistical significance. Heart weight was not different in any group (Fig. S4).
Evaluation of renin and other RAS components
The kidney provides two distinct sources of renin from the juxtaglomerular (JG) cells and the CD. Renin release from JG cells residing in renal cortex is regarded as a rate-limiting step in activation of systemic RAS whereas expression of renin in CD principal cells represents a key component of intrarenal RAS29, 30. Circulating renin is suppressed but urinary and renal medullary renin is enhanced by AngII infusion, highlighting the differences in the response of systemic versus intrarenal RAS20, 31–34. We therefore vigorously evaluated circulating and intrarenal renin status in the current experimental mouse models using multiple approaches including the enzyme activity-based assay, ELISA measurement of prorenin/renin content, and qRT-PCR and immunoblotting analysis of renin expression. Fig. 3A&B and Fig. S5A–D showed renin activity, active renin content, and total renin content from the plasma and urine samples collected from vehicle or AngII infused WT and PRRR279V/L282V mice. All renin parameters were suppressed in plasma samples (Fig. 3A, Fig. S5A&B) but increased in urine samples after AngII treatment (Fig. 3B, Fig. S5C&D). In contrast, PRRR279V/L282V mice exhibited reduced baseline renin level from both plasma and urine, and blunted increase of urinary renin to AngII infusion as compared with WT controls. The pattern changes among the three renin parameters were quite similar.
Figure. 3.
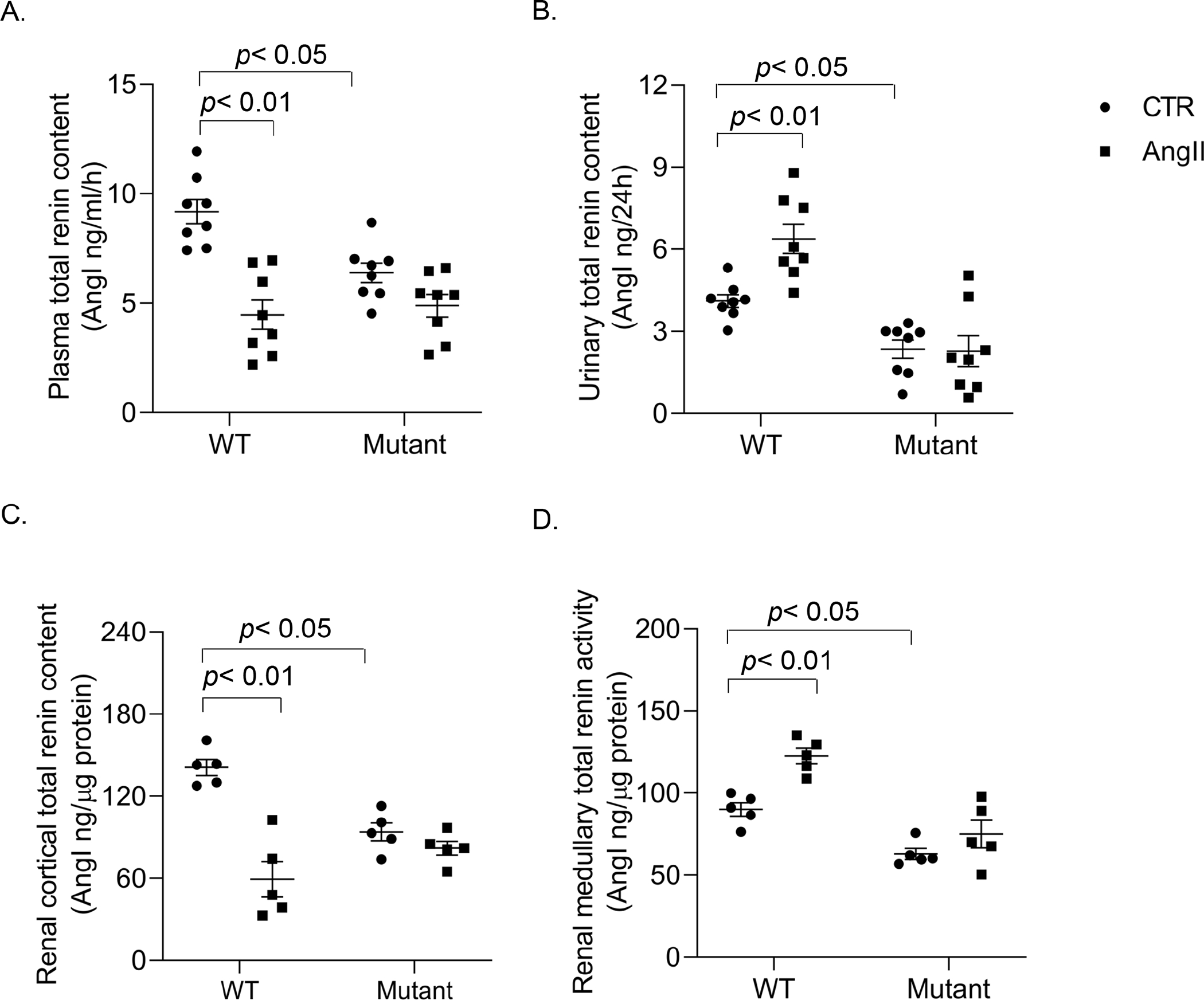
Renin enzyme activity assay on biological fluid and renal samples. Plasma, urine, tissue lysates of the renal cortex and inner medulla samples from WT and PRRR279V/L282V mice following vehicle or AngII treatment were collected at the end of the experiment (day 14), and subjected to renin enzyme activity assay for assessment of total renin content in plasma (A), urine (B), renal cortex (C), and renal medulla (D). The statistical significance was determined by using two-way ANOVA with the Bonferroni test for multiple comparisons, and the p values were indicated in the figure. Data are mean ± SE. WT, wild-type; Mutant, PRRR279V/L282V.
The renin enzyme activity assay was also performed on the renal cortex (Fig. 3C, Fig. S5E&F) and inner medulla (Fig. 3D, Fig. S5G&H) of the two genotypes following vehicle or AngII infusion. Renal cortical and inner medullary renin followed almost exactly the same pattern of changes as plasma and urinary renin, respectively. ELISA was subsequently performed to examine prorenin/renin content from the same renal cortical and inner medullary samples. Of note, this assay was unable to distinguish between prorenin versus renin. Similarly, prorenin/renin content was suppressed in the renal cortex but was stimulated in the renal inner medulla of WT mice following AngII infusion contrasting to the largely blunted renin response in both regions of PRRR279V/L282V mice. Additionally, baseline prorenin and renin content was reduced in PRRR279V/L282V mice as compared with WT controls. (Fig. S6). Lastly, renin mRNA and protein expression in the two renal regions was examined by using qRT-PCR and immunoblotting analysis, respectively. Following AngII infusion, renin expression was downregulated in the renal cortex but upregulated in the inner medulla at both mRNA and protein level in the WT mice. Again, PRRR279V/L282V mice had reduced renin mRNA (Fig. 4A&B) and protein expression (Fig. 4C) at basal condition and the blunted renin expression response following AngII infusion. Together, these results support an essential role of sPRR as a mediator of AngII-induced intrarenal renin expression as well as a key determinant of JG renin release.
Figure. 4.
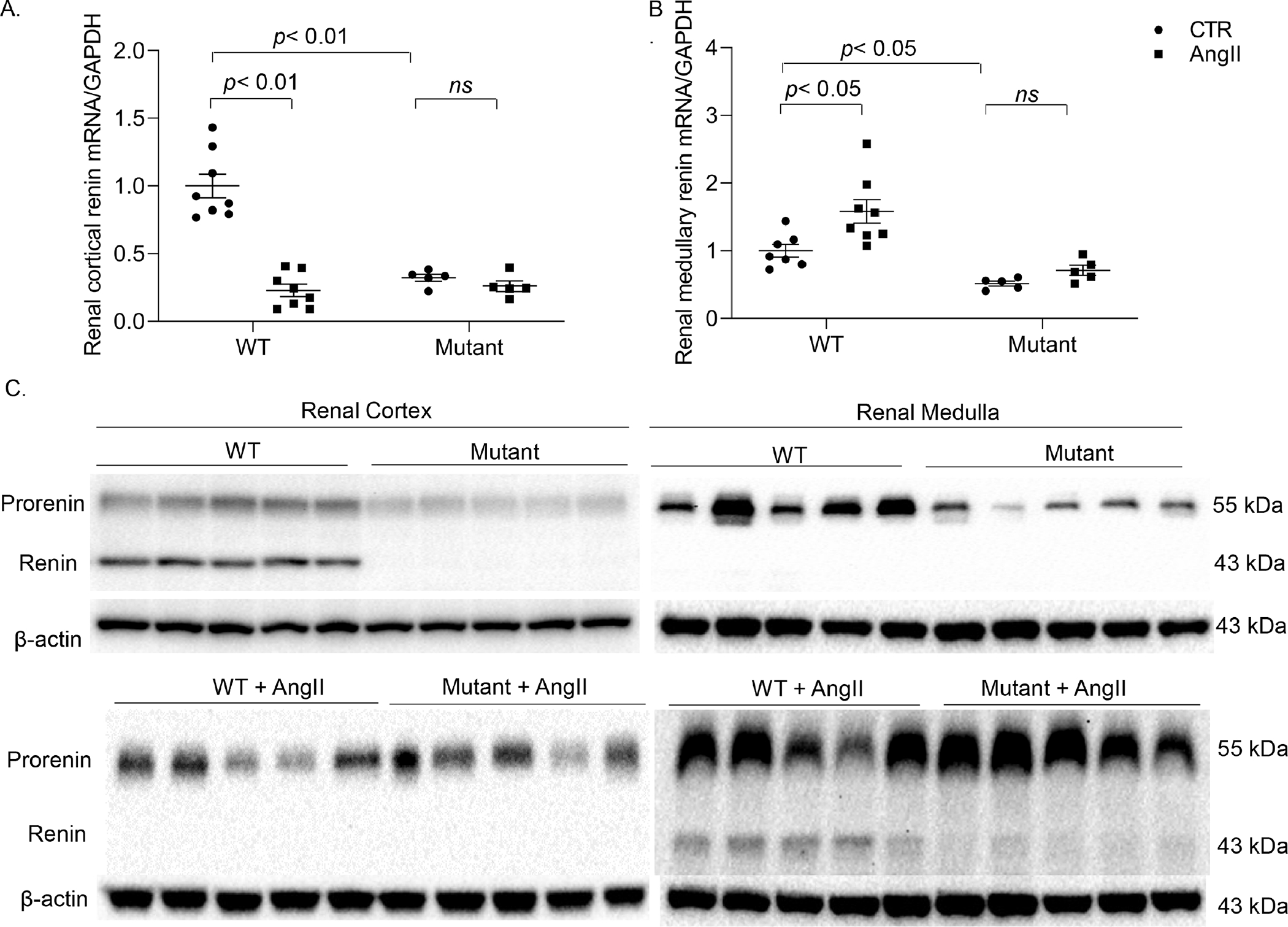
Assessment of renin expression in renal tissues. Tissue lysates of the renal cortex and inner medulla samples from WT and PRRR279V/L282V mice following vehicle or AngII treatment were collected at the end of the experiment (day 14). Renin mRNA expression of renal cortex and inner medulla was determined by qRT-PCR analysis (A&B). Renin protein expression of renal cortex and inner medulla was further validated by immunoblotting analysis (C). N = 5–8 per each group. The statistical significance was determined by using two-way ANOVA with the Bonferroni test for multiple comparisons, and the p values were indicated in the figure. Data are mean ± SE. WT, wild-type; Mutant, PRRR279V/L282V.
Besides above-mentioned renin, we have also assessed the status of several other RAS components including AGT and AngI, as well as aldosterone. In particular, the work from Navar’s group showed that elevated urinary AGT is associated with enhanced intrarenal RAS in animal models of hypertension 35. Interestingly both plasma and urinary AGT in WT mice was elevated by AngII infusion. In contrast, PRRR279V/L282V mice had reduced basal levels of plasma and urinary AGT as well as their blunted response to AngII infusion (Fig. S7 A&B). Similarly, basal plasma aldosterone was lower and its response to AngII was also blunted in PRRR279V/L282V mice (Fig. S7 C). In contrast, plasma and renal cortical AngI levels were suppressed during AngII infusion but urinary AngI excretion and renal medullary AngI generation were significantly increased in WT mice, all of which were blunted in PRRR279V/L282V mice (Fig. S7 D–G). These results provide additional support of sPRR as an important regulator of intrarenal RAS during AngII-induced hypertension as well as the activity of systemic RAS under basal condition.
Evaluation of sodium transporters expression and ENaC activity
We have previously shown that PRR specifically targets renal medullary α-ENaC to mediate the hypertensive response to AngII11, 36. Therefore, we examined the renal expression of ENaC, particularly the αlpha subunit, in PRRR279V/L282V mice following AngII infusion. Renal cortical mRNA expression of all 3 ENaC subunits as assessed by qRT-PCR remained quite constant among various experimental groups (Fig. 5B). In contrast, renal medullary α-ENaC mRNA expression in WT mice was elevated by AngII infusion, which was blunted in PRRR279V/L282V mice (Fig. 5C). The changes in renal medullary α-ENaC was confirmed by immunoblotting analysis (Fig. 5A). Besides ENaC, other Na+ transporters may also be involved in sPRR mediation of the hypertensive response to AngII infusion. In WT mice, AngII infusion selectively elevated renal protein expression of NKCC2 and NCC but not NHE3. Interestingly, PRRR279V/L282V mice had elevated baseline protein expression of NKCC2 and NCC but not NHE3, none of which showed further increases following AngII infusion (Fig. S8).
Figure. 5.
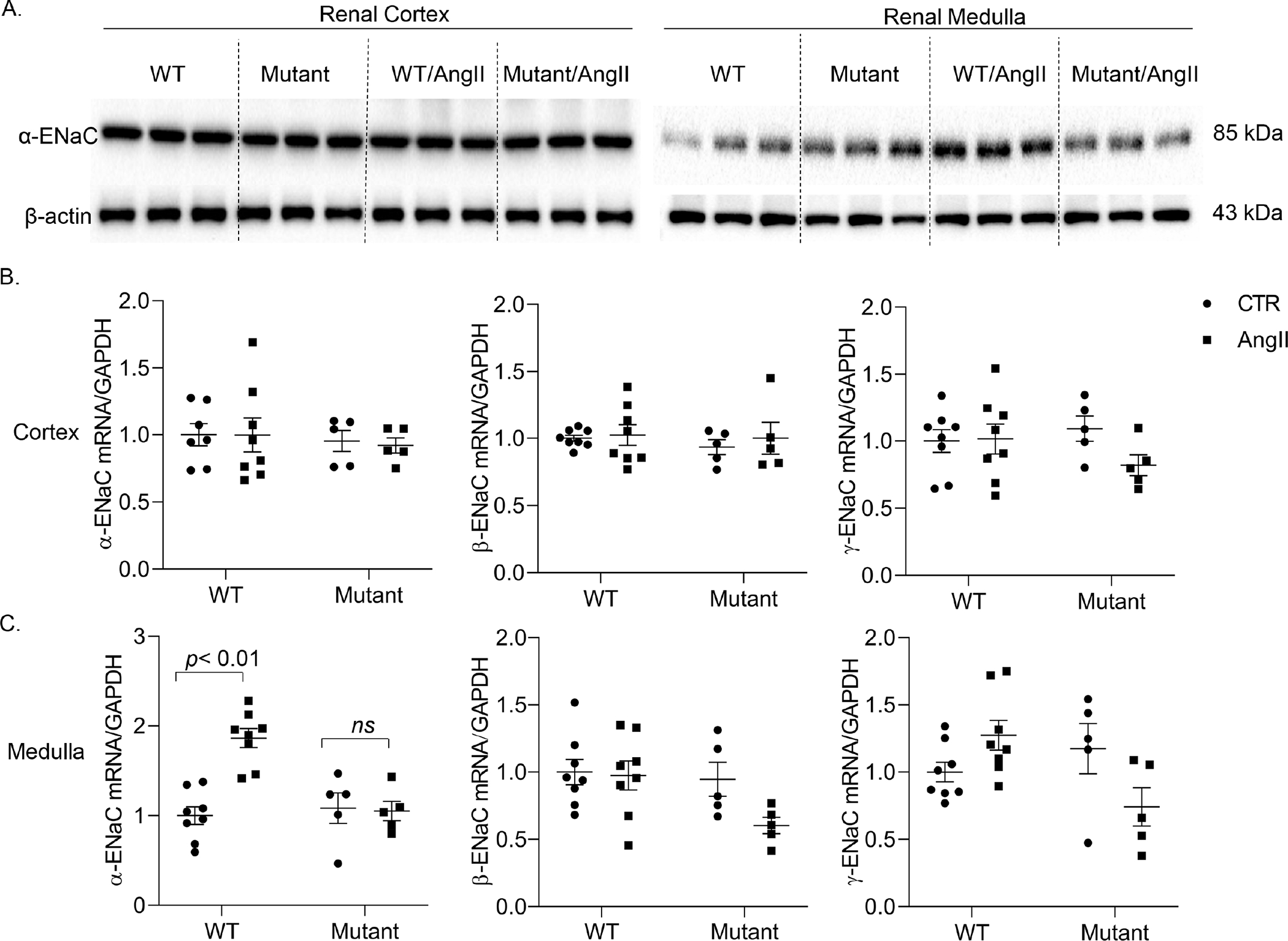
Assessment of renal regional expression of ENaC subunits. mRNA expression of the 3 subunits of ENaC in renal cortex (A) and inner medulla (B) of vehicle or AngII treated WT and PRRR279V/L282V mice was determined by qRT-PCR. The protein expression of α-ENaC in the two regions was verified by immunoblotting analysis (C). The protein expression was normalized by β-actin and the mRNA expression by GAPDH. The statistical significance was determined by using two-way ANOVA with the Bonferroni test for multiple comparisons, and the p values were indicated in the figure. Data are mean ± SE. WT, wild-type; Mutant, PRRR279V/L282V.
Next, to examine the direct role of endogenous sPRR in regulation of ENaC activity in the setting of AngII treatment, we employed Ussing chamber technique to measure amiloride-sensitive short-circuit current25, an index of ENaC activity. Confluent primary culture IMCD cells from WT and PRRR279V/L282V mice were grown on Snapwell membrane and exposed for 24 h to vehicle or AngII. In response to AngII treatment, ENaC activity was elevated in WT but not mutant cells although the baseline values were quite comparable between the two types of cells (Fig. S9). These results support the concept that endogenous sPRR selectively target renal medullary α-ENaC to regulate ENaC activity during AngII treatment.
The direct role of sPRR in renin regulation.
The in vivo studies suggest a renin regulatory role of sPRR both at juxtaglomerular cells (JG cells) and the CD. To gain evidence of the direct role of sPRR in renin regulation at the two locations, we performed in vitro experiments to examine the effect of sPRR on renin expression in As4.1 cells, a renin-expressing cell line isolated from mouse renal tumor and M1 cells, a mouse cell line derived from the CD. We further tested if β-catenin signaling was responsible for the renin-regulatory role of sPRR. Therefore, after reaching confluence, the cells were exposed to 10 nM sPRR-His for 24 h in the presence of absence of a β-catenin signaling inhibitor ICG-001, followed by assessment of various aspects of renin by the enzyme activity-based assay, ELISA measurement of prorenin/renin content, and qRT-PCR analysis of renin mRNA expression. The exposure to sPRR-His consistently elevated renin activity, active renin content, and total renin content, prorenin/renin content, and renin mRNA expression, all of which were sensitive to inhibition by ICG-001 (Fig. 6, Fig. S10). The pattern of changes was not different between the two cell types. These results demonstrated that sPRR stimulated renin expression in both As4.1 and M1 cells through activation of β-catenin signaling.
Figure. 6.
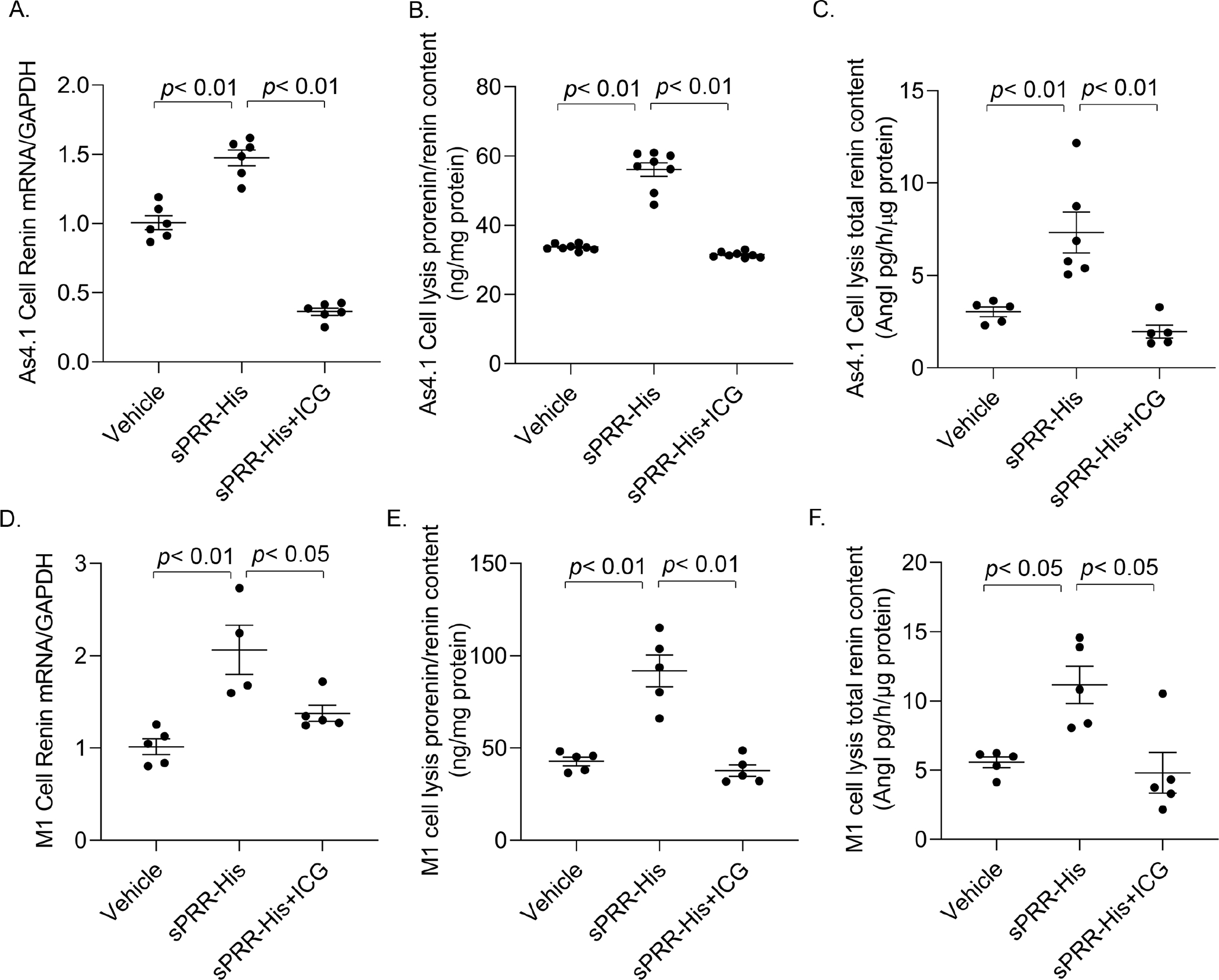
The role and mechanism of sPRR in regulation of renin expression in cultured As4.1 and M-1 cells. The confluent cells were pretreated for 1 h with the β-catenin inhibitor ICG-001 at 10 μM and then treated for 24h with 10 nM sPRR-His. At the end of the experiment, cells and/or the media were harvested for qRT-PCR analysis of renin mRNA, enzyme activity-based assay for total renin content, and ELISA measurement of prorenin/renin content. (A) Renin mRNA expression in As4.1 cells. (B) Cellular total prorenin/renin content in As4.1 cells. (C) Cellular total renin content in As4.1 cells. (D) Renin mRNA expression in M-1 cells. (E) Cellular total prorenin/renin content in M-1 cells. (F) Cellular total renin content in M-1 cells. N = 5–8 per group. Data are mean ± SE.
Discussion
In the present study, we employed CRISPR/Cas9 strategy to generate mice with mmutagenesis of the overlapping cleavage site for both S1P and furin in PRR. PRRR279V/L282V mice showed a 53% reduction of plasma sPRR and an 82% reduction of renal sPRR. Radiotelemetry demonstrated that these mice exhibited blunted hypertensive response to AngII infusion accompanied with a widespread suppression of renin level and a selective reduction of renal medullary α-ENaC expression. Furthermore, primary IMCD cells isolated from PRRR279V/L282V mice showed attenuation of AngII-induced ENaC activity as assessed by using Ussing chamber technique.
The recent identification of S1P as a predominant PRR cleavage protease from two independent laboratories offers a unique opportunity to investigate the function of endogenous sPRR 6, 7. In this regard, pharmacological inhibition and nephron-specific deletion of S1P contribute to understanding the role of S1P-derived sPRR in determining urine concentrating capability 22 as well as the BP response to AngII infusion 37. However, this approach is limited in that besides PRR, S1P has a number of other known substrates including membrane-bound latent transcription factors sterol regulatory element-binding transcription factor (SREBP) and membrane-bound activating transcription factor 6 (ATF6). To overcome this limitation, mutagenesis of PRR cleavage site will allow precise manipulation of the cleavage process for PRR but not other substrates. In the present study, we have successfully employed CRISP/Cas9 strategy to generate mice with mutagenesis of PRR cleavage site as validated by PCR genotyping coupled with restriction enzyme digestion and direct sequencing. It is interesting to note that despite the systemic mutagenesis approach the reduction of renal sPRR in PRRR279V/L282V mice appears much greater than that in the circulation, raising a possibility that the cleavage efficiency may vary depending on the type of tissues. Our result is compatible with the concept that S1P may function more effectively in the kidney than in other tissues based on the assumption that furin may play a minimal role in the cleavage process 7. On the other hand, the possibility of tissue-specific action of the proteases is raised by Morosin et al. who report that the generation of sPRR from placental trophoblasts is unaffected by inhibition of either furin or S1P, suggesting a novel enzymatic source of sPRR in the placenta 38.
A majority of previous studies have demonstrated an essential role of renal PRR in mediating of AngII-induced hypertension 9, 39, 40. The prohypertensive action of PRR is further mapped to the CD, an important nephron site for fine-tuning urinary Na+ excretion 39. A clue suggesting involvement of sPRR came from early evidence that renal abundance and urinary excretion of sPRR were elevated during AngII infusion 41. Unfortunately we were unable to provide urinary sPRR data since for some unknown reason the recent batches of ELISA kit from IBL International, the only source of this type of kit, can no longer detect mouse urinary sPRR as it used to although the kit still works on plasma samples. Along this line, we have recently reported that the increase of renal PRR protein expression in response to AngII infusion took place selectively in the renal medulla but not renal cortex, providing the substrate for renal medullary generation of sPRR 37. At the functional level, S1P inhibition with PF effectively attenuated AngII-induced hypertension accompanied by reduced sPRR production 37. The current study has made a significant extension of these observations by creation and characterization of a novel mouse model of mutagenesis of the cleavage site of PRR. As expected, PRRR279V/L282V mice exhibited a blunted hypertensive response to AngII infusion, establishing requirement of sPRR for the hypertension development.
Existence of intrarenal RAS is highlighted by the expression of renin in the renal medulla, particularly the CD 42–44, where the expression is increased by AngII infusion 29, 45, contrasting to suppressed plasma renin under this condition. Overactivation of intrarenal RAS including increased CD renin expression contributes to AngII-induced hypertension46–49. Typically, in response to AngII infusion, urinary and renal medullary renin is elevated, an index of activated intrarenal RAS whereas circulating and renal cortical renin is suppressed as a result of the negative feedback response of systemic RAS. In the present study, we performed vigorous evaluation of renin status including renin activity assay, ELISA determination of prorenin/renin content, and qRT-PCR and immunoblotting analysis of renin expression in the AngII infused PRRR279V/L282V mice. Clear-cut results showed that AngII-induced increases in renin activity and expression in the urine and renal medulla were all consistently blunted in PRRR279V/L282V mice, supporting an essential role of sPRR in determining the local renin response to AngII infusion. The assessment of renin status of PRRR279V/L282V mice further showed that besides the alteration of AngII-induced renal medullary renin response, a drastic change was also observed in the basal level of circulating renin, an important indicator of systemic RAS. In PRRR279V/L282V mice, the basal level of renin activity and renin content in the plasma and renal cortex was consistently suppressed in PRRR279V/L282V mice as compared with WT controls. qRT-PCR and immunoblotting analysis further showed suppressed renal renin mRNA and protein expression. These results strongly suggest that sPRR may function as a master regulator of basal renin expression irrespective of the location, namely the juxtaglomerular (JG) cells or the CD. In support of this notion, in vitro evidence demonstrated a direct stimulatory effect of sPRR-His on renin expression in both As4.1 and M1 cells. Indeed, salt depletion, a well-known stimulus of JG renin response, induces renal PRR expression 50. PRR is expressed in the macula densa (MD) and MD-specific deletion of PRR attenuated renin secretion from the JG cells 51. It remains elusive whether the renin-regulatory role of MD PRR is mediated by sPRR whereby sPRR acts in a paracrine manner to mediate the communication between MD and JG cells for the control of renin secretion. Additionally, sPRR seems to regulate the release of aldosterone in addition to renin. In this regard, plasma aldosterone in PRRR279V/L282V mice was suppressed at baseline as well as during AngII infusion, suggesting a potential role of sPRR in the control of aldosterone secretion, possibly from adrenal glands. In support of this notion, among multiple tissues tested, the highest amount of sPRR was detected in adrenal glands. Moreover, expression of PRR is elevated in tumor tissues in patients with aldosterone-producing adenomors (APAs) 52.
In a classical view, PRR or sPRR is capable of binding prorenin and renin to increase their catalytic activity through the conformational change of the substrates 1. It is intriguing that besides renin activity, other renin parameters such as active renin content, total renin content, and renin mRNA and protein expression in the kidney were all blunted in PRRR279V/L282V mice under basal condition or following AngII infusion, in parallel with the suppressed aldosterone and AngI levels. The broad suppression of multiple renin parameters were also seen following intramedullary infusion of PRO20 in rats 53 or CD-specific deletion of PRR 11. Together, these results suggest that sPRR-dependent regulation of renin goes much beyond catalytic activity. Future studies are needed to examine the molecular mechanism of how sPRR regulates renin at multiple levels. Irrespective of the underlying mechanism, the present study has firmly established the relationship between sPRR and renin. This evidence is timely given the long debate concerning the primary function of PRR as a regulator of the RAS versus lysosome/autophagy or both 14, 21.
Within the CD, most Na+ transport is thought to occur through ENaC in the CCD 54, in agreement with the observation that AngII infusion activates renal cortical ENaC 55. However, a series of our studies including the present one demonstrated that renal medullary expression of α-ENaC but not β- or γ-subunit was elevated during AngII infusion, which was blunted by renal medullary PRR antagonism 53, CD-specific deletion of PRR 39. In agreement with our findings, Quadri et al. showed that adenoviral-mediated delivery of shRNA against PRR to the rat kidney selectively reduced renal medullary expression of α-ENaC but not the β or γ subunit 56. In cultured mpkCCD cells, administration of sPRR acutely stimulated ENaC activity via Nox-4-derived H2O2 and chronically activates transcription of α-ENaC via β-catenin signaling 25. In agreement with these findings, the present study showed that renal medullary mRNA and protein expression of α-ENaC was elevated by AngII infusion in WT controls, which was blunted in PRRR279V/L282V mice. It seems reasonable to speculate that sPRR-mediated activation of renal medullary α-ENaC may in part contribute to AngII-induced hypertension. As shown previously 55, renal abundances of Na+ transporters in TAL and DCT, namely NKCC2 and NCC, were both elevated by AngII infusion in WT mice. Interestingly, PRRR279V/L282V mice had increased basal levels of NKCC2 and NCC, rendering them insensitive to AngII infusion and thus arguing their role in the hypertension development. The mechanism of altered basal expression of NKCC2 and NCC in PRRR279V/L282V mice remains unknown but may suggest a direct inhibitory effect of sPRR on these Na+ transporters or reflect a compensatory response to suppressed activity of RAS.
Supplementary Material
Figure. 2.
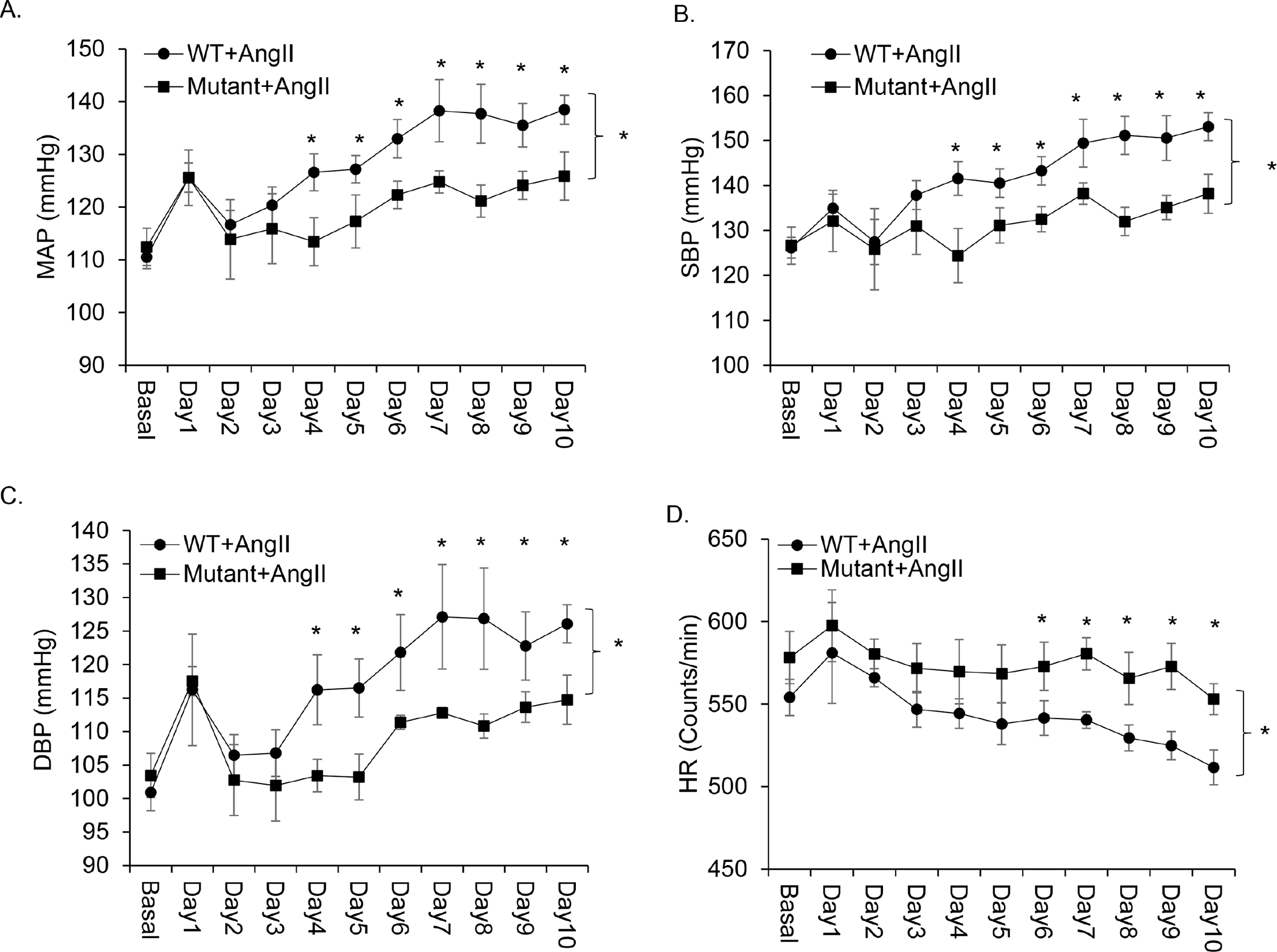
Role of endogenous sPRR in AngII-induced hypertension. WT and PRRR279V/L282V mice were instrumented with radiotelemetric devices and treated with vehicle or AngII (n = 8 per each group). (A) Mean arterial blood pressure (MAP), (B) Systolic blood pressure (SBP), (C) Diastolic blood pressure (DBP), and (D) Heart rate (HR) over the first 10 days of AngII infusion. The statistical significance was determined by using two-way ANOVA with repeated measurements. *, p <0.05 vs. WT + AngII group. Data are mean ± SE. WT, wild-type; Mutant, PRRR279V/L282V.
Novelty and Significance.
What Is New?
sPRR is not only a disease biomarker but also exerts biological functions in a variety of physio-pathological conditions including hypertension.
S1P is reported as a predominant source of endogenous sPRR production while furin may play a contributable role.
Here we generated a novel mouse model with mutagenesis of the cleavage site of PRR to provide definitive evidence for the function of sPRR in BP regulation.
What Is Relevant?
RAS inhibition has been a cornerstone therapy for hypertension and cardiovascular and renal disease for more than half of century.
Despite a century research, our understanding of this system is quite primitive.
PRR was cloned more than 10 years ago but it’s relationship with RAS has been debated.
A better understanding of the cleavage processes for PRR may offer a new therapeutic target for management of hypertension.
Perspectives.
The prevalence of hypertension is increasing with nearly 1 out of 2 adults in the United States suffering from this disease and the BP being uncontrolled for most of the cases 57. Importance of the RAS in hypertension is highlighted by the wide use of RAS inhibitors as a cornerstone antihypertensive therapy. However, our understanding of the RAS is still primitive. RAS inhibition with ACE inhibitors or AT1R blockers is associated with a number of limitations, particularly overproduction of prorenin/renin (hyperreninemia) which may induce tissue damage via RAS-dependent or independent mechanisms. Although PRR was originally cloned as a specific receptor for prorenin and renin, its relationship with the RAS has been debated. The present study for the first time generated a novel mouse model of mutagenesis of the cleavage site of PRR which abrogates the generation of sPRR. The phenotype of this mouse model has been extensively characterized during AngII-induced hypertension. Compelling evidence from this study supports endogenous sPRR as a master regulator of renin expression and thus the hypertensive response to AngII. This evidence strongly supports the clarification of PRR/sPRR as an integrative member of the RAS as well as the continued use of its current name but not ATP6ap2 at least in the context of the RAS and hypertension. Our results suggest that targeting sPRR may offer an effective antihypertensive therapy without hyperreninemia which can not be achieved with the existing anti-RAS therapies.
Sources of Funding
This work was supported by National Institutes of Health Grants HL139689, DK104072, HL135851, and VA Merit Review from the Department of Veterans Affairs. T. Yang is Senior Research Career Scientist in Department of Veterans Affairs.
Footnotes
Conflict of Interest
None.
References
- 1.Nguyen G, Delarue F, Burckle C, Bouzhir L, Giller T and Sraer JD. Pivotal role of the renin/prorenin receptor in angiotensin II production and cellular responses to renin. The Journal of clinical investigation. 2002;109:1417–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Burckle C and Bader M. Prorenin and its ancient receptor. Hypertension. 2006;48:549–51. [DOI] [PubMed] [Google Scholar]
- 3.Ludwig J, Kerscher S, Brandt U, Pfeiffer K, Getlawi F, Apps DK and Schagger H. Identification and characterization of a novel 9.2-kDa membrane sector-associated protein of vacuolar proton-ATPase from chromaffin granules. The Journal of biological chemistry. 1998;273:10939–47. [DOI] [PubMed] [Google Scholar]
- 4.Cousin C, Bracquart D, Contrepas A, Corvol P, Muller L and Nguyen G. Soluble form of the (pro)renin receptor generated by intracellular cleavage by furin is secreted in plasma. Hypertension. 2009;53:1077–82. [DOI] [PubMed] [Google Scholar]
- 5.Yoshikawa A, Aizaki Y, Kusano K, Kishi F, Susumu T, Iida S, Ishiura S, Nishimura S, Shichiri M and Senbonmatsu T. The (pro)renin receptor is cleaved by ADAM19 in the Golgi leading to its secretion into extracellular space. Hypertension research : official journal of the Japanese Society of Hypertension. 2011;34:599–605. [DOI] [PubMed] [Google Scholar]
- 6.Nakagawa T, Suzuki-Nakagawa C, Watanabe A, Asami E, Matsumoto M, Nakano M, Ebihara A, Uddin MN and Suzuki F. Site-1 protease is required for the generation of soluble (pro)renin receptor. J Biochem. 2017;161:369–379. [DOI] [PubMed] [Google Scholar]
- 7.Fang H, Xu C, Lu A, Zou CJ, Xie S, Chen Y, Zhou L, Liu M, Wang L, Wang W and Yang T. (Pro)renin receptor mediates albumin-induced cellular responses: role of site-1 protease-derived soluble (pro)renin receptor in renal epithelial cells. American journal of physiology Cell physiology. 2017;313:C632–C643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Advani A, Kelly DJ, Cox AJ, White KE, Advani SL, Thai K, Connelly KA, Yuen D, Trogadis J, Herzenberg AM, Kuliszewski MA, Leong-Poi H and Gilbert RE. The (Pro)renin receptor: site-specific and functional linkage to the vacuolar H+-ATPase in the kidney. Hypertension. 2009;54:261–9. [DOI] [PubMed] [Google Scholar]
- 9.Ramkumar N, Stuart D, Mironova E, Bugay V, Wang S, Abraham N, Ichihara A, Stockand JD and Kohan DE. Renal tubular epithelial cell prorenin receptor regulates blood pressure and sodium transport. American journal of physiology Renal physiology. 2016:ajprenal 00088 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Prieto MC, Reverte V, Mamenko M, Kuczeriszka M, Veiras LC, Rosales CB, McLellan M, Gentile O, Jensen VB, Ichihara A, McDonough AA, Pochynyuk OM and Gonzalez AA. Collecting duct prorenin receptor knockout reduces renal function, increases sodium excretion, and mitigates renal responses in ANG II-induced hypertensive mice. American journal of physiology Renal physiology. 2017;313:F1243–F1253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Peng K, Lu X, Wang F, Nau A, Chen R, Zhou SF and Yang T. Collecting Duct (Pro)Renin Receptor Targets ENaC to Mediate Angiotensin II-Induced Hypertension. American journal of physiology Renal physiology. 2016:ajprenal 00178 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kinouchi K, Ichihara A, Sano M, Sun-Wada GH, Wada Y, Kurauchi-Mito A, Bokuda K, Narita T, Oshima Y, Sakoda M, Tamai Y, Sato H, Fukuda K and Itoh H. The (pro)renin receptor/ATP6AP2 is essential for vacuolar H+-ATPase assembly in murine cardiomyocytes. Circulation research. 2010;107:30–4. [DOI] [PubMed] [Google Scholar]
- 13.Oshima Y, Morimoto S and Ichihara A. Roles of the (pro)renin receptor in the kidney. World journal of nephrology. 2014;3:302–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ichihara A and Yatabe MS. The (pro)renin receptor in health and disease. Nat Rev Nephrol. 2019;15:693–712. [DOI] [PubMed] [Google Scholar]
- 15.Zhu Q and Yang T. Enzymatic sources and physio-pathological functions of soluble (pro)renin receptor. Current opinion in nephrology and hypertension. 2018;27:77–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ichihara A, Kaneshiro Y, Takemitsu T, Sakoda M and Itoh H. The (pro)renin receptor and the kidney. Seminars in nephrology. 2007;27:524–8. [DOI] [PubMed] [Google Scholar]
- 17.Ichihara A, Sakoda M, Kurauchi-Mito A, Kaneshiro Y and Itoh H. Involvement of (pro)renin receptor in the glomerular filtration barrier. Journal of molecular medicine. 2008;86:629–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gonzalez AA, Lara LS, Luffman C, Seth DM and Prieto MC. Soluble form of the (pro)renin receptor is augmented in the collecting duct and urine of chronic angiotensin II-dependent hypertensive rats. Hypertension. 2011;57:859–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wang F, Lu X, Peng K, Du Y, Zhou SF, Zhang A and Yang T. Prostaglandin E-Prostanoid4 Receptor Mediates Angiotensin II-Induced (Pro)Renin Receptor Expression in the Rat Renal Medulla. Hypertension. 2014;64:369–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wang F, Lu X, Liu M, Feng Y, Zhou SF and Yang T. Renal medullary (pro)renin receptor contributes to angiotensin II-induced hypertension in rats via activation of the local renin-angiotensin system. BMC medicine. 2015;13:278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sun Y, Danser AHJ and Lu X. (Pro)renin receptor as a therapeutic target for the treatment of cardiovascular diseases? Pharmacol Res. 2017;125:48–56. [DOI] [PubMed] [Google Scholar]
- 22.Wang F, Xu C, Luo R, Peng K, Ramkumar N, Xie S, Lu X, Zhao L, Zuo CJ, Kohan DE and Yang T. Site-1 protease-derived soluble (pro)renin receptor targets vasopressin receptor 2 to enhance urine concentrating capability. JCI Insight. 2019;4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sakai J, Rawson RB, Espenshade PJ, Cheng D, Seegmiller AC, Goldstein JL and Brown MS. Molecular identification of the sterol-regulated luminal protease that cleaves SREBPs and controls lipid composition of animal cells. Mol Cell. 1998;2:505–14. [DOI] [PubMed] [Google Scholar]
- 24.Suzuki F, Nakagawa T, Kakidachi H, Murakami K, Inagami T and Nakamura Y. The dominant role of the prosegment of prorenin in determining the rate of activation by acid or trypsin: studies with molecular chimeras. Biochemical and biophysical research communications. 2000;267:577–80. [DOI] [PubMed] [Google Scholar]
- 25.Wang F, Luo R, Peng K, Liu X, Xu C, Lu X, Soodvilai S and Yang T. Soluble (pro)renin receptor regulation of ENaC involved in aldosterone signaling in cultured collecting duct cells. American journal of physiology Renal physiology. 2020;318:F817–F825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Soodvilai S, Jia Z and Yang T. Hydrogen peroxide stimulates chloride secretion in primary inner medullary collecting duct cells via mPGES-1-derived PGE2. American journal of physiology Renal physiology. 2007;293:F1571–6. [DOI] [PubMed] [Google Scholar]
- 27.Fang H, Xu C, Lu A, Zou CJ, Xie S, Chen Y, Zhou L, Liu M, Wang L, Wang W and Yang T. (Pro) renin receptor mediates albumin-induced cellular responses: role of site-1 protease-derived soluble (pro) renin receptor in renal epithelial cells (vol 313, pg C632, 2017). Am J Physiol-Cell Ph 2018;314:C255–C255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wang F, Luo R, Zou CJ, Xie S, Peng K, Zhao L, Yang KT, Xu C and Yang T. Soluble (pro)renin receptor treats metabolic syndrome in mice with diet-induced obesity via interaction with PPARgamma. JCI Insight. 2020;5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Prieto-Carrasquero MC, Botros FT, Kobori H and Navar LG. Collecting Duct Renin: A major player in Angiotensin II-dependent Hypertension. J Am Soc Hypertens. 2009;3:96–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gonzalez AA, Liu L, Lara LS, Seth DM, Navar LG and Prieto MC. Angiotensin II stimulates renin in inner medullary collecting duct cells via protein kinase C and independent of epithelial sodium channel and mineralocorticoid receptor activity. Hypertension. 2011;57:594–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wang F, Lu X, Peng K, Zhou L, Li C, Wang W, Yu X, Kohan DE, Zhou SF and Yang T. COX-2 Mediates Angiotensin II-Induced (Pro)Renin Receptor Expression in the Rat Renal Medulla. American journal of physiology Renal physiology. 2014;307:F25–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kobori H, Nangaku M, Navar LG and Nishiyama A. The intrarenal renin-angiotensin system: from physiology to the pathobiology of hypertension and kidney disease. Pharmacological reviews. 2007;59:251–87. [DOI] [PubMed] [Google Scholar]
- 33.Yang T. Crosstalk between (Pro)renin receptor and COX-2 in the renal medulla during angiotensin II-induced hypertension. Current opinion in pharmacology. 2015;21:89–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yang T and Xu C. Physiology and Pathophysiology of the Intrarenal Renin-Angiotensin System: An Update. Journal of the American Society of Nephrology : JASN. 2017;28:1040–1049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Shao W, Miyata K, Katsurada A, Satou R, Seth DM, Rosales CB, Prieto MC, Mitchell KD and Navar LG. Increased angiotensinogen expression, urinary angiotensinogen excretion, and tissue injury in nonclipped kidneys of two-kidney, one-clip hypertensive rats. American journal of physiology Renal physiology. 2016;311:F278–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wang F, Luo R, Peng K, Liu X, Xu C, Lu X, Soodvilai S and Yang T. Soluble (Pro)Renin Receptor Regulation of ENaC Involved in Aldosterone Signaling in Cultured Collecting Duct Cells. American journal of physiology Renal physiology. 2020;318: F817–F825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Feng Y, Peng K, Luo R, Wang F and Yang T. Site-1 Protease-Derived Soluble (Pro)Renin Receptor Contributes to Angiotensin II-Induced Hypertension in Mice. Hypertension. 2021;77:405–416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Morosin SK, Delforce SJ, Lumbers ER and Pringle KG. Cleavage of the soluble (pro)renin receptor (sATP6AP2) in the placenta. Placenta. 2020;101:49–56. [DOI] [PubMed] [Google Scholar]
- 39.Peng K, Lu X, Wang F, Nau A, Chen R, Zhou SF and Yang T. Collecting duct (pro)renin receptor targets ENaC to mediate angiotensin II-induced hypertension. American journal of physiology Renal physiology. 2017;312:F245–F253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gonzalez AA and Prieto MC. Roles of collecting duct renin and (pro)renin receptor in hypertension: mini review. Ther Adv Cardiovasc Dis. 2015;9:191–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gonzalez AA, Lara LS, Luffman C, Seth DM and MC P. Soluble form of the (pro)renin receptor is augmented in the collecting duct and urine of chronic angiotensin II-dependent hypertensive rats. Hypertension. 2011:57(4): 859–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ingelfinger JR, Pratt RE, Ellison K and Dzau VJ. Sodium regulation of angiotensinogen mRNA expression in rat kidney cortex and medulla. The Journal of clinical investigation. 1986;78:1311–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Rohrwasser A, Morgan T, Dillon HF, Zhao L, Callaway CW, Hillas E, Zhang S, Cheng T, Inagami T, Ward K, Terreros DA and Lalouel JM. Elements of a paracrine tubular renin-angiotensin system along the entire nephron. Hypertension. 1999;34:1265–74. [DOI] [PubMed] [Google Scholar]
- 44.Kang JJ, Toma I, Sipos A, Meer EJ, Vargas SL and Peti-Peterdi J. The collecting duct is the major source of prorenin in diabetes. Hypertension. 2008;51:1597–604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Prieto-Carrasquero MC, Harrison-Bernard LM, Kobori H, Ozawa Y, Hering-Smith KS, Hamm LL and Navar LG. Enhancement of collecting duct renin in angiotensin II-dependent hypertensive rats. Hypertension. 2004;44:223–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Gonzalez-Villalobos RA, Janjoulia T, Fletcher NK, Giani JF, Nguyen MT, Riquier-Brison AD, Seth DM, Fuchs S, Eladari D, Picard N, Bachmann S, Delpire E, Peti-Peterdi J, Navar LG, Bernstein KE and McDonough AA. The absence of intrarenal ACE protects against hypertension. The Journal of clinical investigation. 2013;123:2011–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ramkumar N, Ying J, Stuart D and Kohan DE. Overexpression of Renin in the collecting duct causes elevated blood pressure. American journal of hypertension. 2013;26:965–72. [DOI] [PubMed] [Google Scholar]
- 48.Ramkumar N and Kohan DE. Role of collecting duct renin in blood pressure regulation. Am J Physiol Regul Integr Comp Physiol. 2013;305:R92–4. [DOI] [PubMed] [Google Scholar]
- 49.Ramkumar N, Stuart D, Rees S, Hoek AV, Sigmund CD and Kohan DE. Collecting duct-specific knockout of renin attenuates angiotensin II-induced hypertension. American journal of physiology Renal physiology. 2014;307:F931–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Matavelli LC, Huang J and Siragy HM. In vivo regulation of renal expression of (pro)renin receptor by a low-sodium diet. American journal of physiology Renal physiology. 2012;303:F1652–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Riquier-Brison ADM, Sipos A, Prokai A, Vargas SL, Toma L, Meer EJ, Villanueva KG, Chen JCM, Gyarmati G, Yih C, Tang E, Nadim B, Pendekanti S, Garrelds IM, Nguyen G, Danser AHJ and Peti-Peterdi J. The macula densa prorenin receptor is essential in renin release and blood pressure control. American journal of physiology Renal physiology. 2018;315:F521–F534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Yamamoto H, Kaneko K, Ohba K, Morimoto R, Hirose T, Satoh F, Totsune K and Takahashi K. Increased expression of (pro)renin receptor in aldosterone-producing adenomas. Peptides. 2013;49:68–73. [DOI] [PubMed] [Google Scholar]
- 53.Wang F, Lu X, Liu M, Feng Y, Zhou SF and T Y. Renal medullary (pro)renin receptor contributes to angiotensin II-induced hypertension in rats via activation of the local renin-angiotensin system. BMC medicine. 2015:13: 278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Palmer LG, Patel A and Frindt G. Regulation and dysregulation of epithelial Na+ channels. Clinical and experimental nephrology. 2012;16:35–43. [DOI] [PubMed] [Google Scholar]
- 55.Nguyen MT, Han J, Ralph DL, Veiras LC and McDonough AA. Short-term nonpressor angiotensin II infusion stimulates sodium transporters in proximal tubule and distal nephron. Physiol Rep. 2015;3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Quadri S and Siragy HM. (Pro)renin receptor contributes to regulation of renal epithelial sodium channel. Journal of hypertension. 2016;34:486–94; discussion 494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ritchey MD, Gillespie C, Wozniak G, Shay CM, Thompson-Paul AM, Loustalot F and Hong Y. Potential need for expanded pharmacologic treatment and lifestyle modification services under the 2017 ACC/AHA Hypertension Guideline. J Clin Hypertens (Greenwich). 2018;20:1377–1391. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.