Summary
Clinical characteristics.
Variegate porphyria (VP) is both a cutaneous porphyria (with chronic blistering skin lesions) and an acute porphyria (with severe episodic neurovisceral symptoms). The most common manifestation of VP is adult-onset cutaneous blistering lesions (subepidermal vesicles, bullae, and erosions that crust over and heal slowly) of sun-exposed skin, especially the hands and face. Other chronic skin findings include milia, scarring, thickening, and areas of decreased and increased skin pigmentation. Facial hyperpigmentation and hypertrichosis may occur. Cutaneous manifestations may improve in winter and be less prevalent in northern regions and in dark-skinned individuals. Acute neurovisceral symptoms can occur any time after puberty, but less often in the elderly. Acute manifestations are highly variable, but may be similar from episode to episode in a person with recurrent attacks; not all manifestations are present in a single episode; and acute symptoms may become chronic. Symptoms are more common in women than men. The most common manifestations are abdominal pain; constipation; pain in the back, chest, and extremities; anxiety; seizures; and a primarily motor neuropathy resulting in muscle weakness that may progress to quadriparesis and respiratory paralysis. Psychiatric disturbances and autonomic neuropathy can also be observed. Acute attacks may be severe and are potentially fatal.
Diagnosis/testing.
The biochemical diagnosis of VP is established in an individual with elevated urine porphobilinogen (PBG) or porphyrins and a fluorescence peak at ~626 nm on plasma fluorescence scanning; fecal porphyrins are also elevated, with a predominance of coproporphyrin III and protoporphyrin. The molecular diagnosis of VP is established by identification of a heterozygous pathogenic variant in PPOX on molecular genetic testing.
Management.
Treatment of manifestations: The first step in treating either acute neurovisceral attacks or cutaneous manifestations is to identify and remove exacerbating factors (see Agents/circumstances to avoid). Most acute neurovisceral attacks require hospital admission; the presence of seizures, motor neuropathy, and hyponatremia suggest severe disease that ideally should be managed in an ICU. Narcotic analgesics are usually required for pain. Ondansetron or a related drug can be used for nausea and vomiting; phenothiazines can be effective for nausea, agitation, and hallucinations.
Although mild attacks (without seizures, weakness, or hyponatremia and not requiring narcotics) can sometimes be treated in an outpatient setting with glucose loading, most attacks require treatment with intravenous hemin and in-patient observation for additional supportive management.
Cutaneous manifestations are best managed by wearing protective clothing and avoiding exposure to sunlight. Symptoms may decrease when exacerbating factors are removed. No treatment is known to be effective in lowering porphyrin levels and reducing cutaneous symptoms. Analgesics may be needed for painful lesions and antibiotics for superimposed infection.
Prevention of primary manifestations: Acute neurovisceral attacks are less likely to occur if exacerbating factors are corrected or avoided. Recurrent premenstrual acute attacks can be prevented with gonadotropin-releasing hormone analogs; weekly or biweekly hemin infusions to prevent frequent noncyclical attacks may be effective, but experience is lacking. Prevention of the skin manifestations requires protection from sunlight.
Surveillance: Liver imaging at six-month intervals beginning at age 50 years in those who have experienced persistent elevations in porphobilinogen or porphyrins may detect early hepatocellular carcinoma.
Agents/circumstances to avoid: Exacerbating factors that should be avoided include drugs such as: barbiturates, sulfonamide antibiotics, griseofulvin, rifampin, most anticonvulsants including phenytoin and carbamazepine, alcohol, ergot alkaloids, metoclopramide, and progestins. Although birth control pills should generally be avoided, low-dose hormonal preparations may be tolerated. Concomitant illnesses should be treated effectively using drugs that are considered safe whenever possible. Updated lists of safe and unsafe drugs are maintained at the websites of the American Porphyria Foundation and the European Porphyria Network.
Evaluation of relatives at risk: At-risk family members can be offered molecular genetic testing for the family-specific PPOX pathogenic variant to identify those who are heterozygous (for the purpose of counseling regarding appropriate use of drugs and avoidance of known exacerbating factors). While biochemical testing, especially plasma fluorescence scanning and fecal porphyrin analysis, is also useful, it is less sensitive than molecular genetic testing.
Pregnancy management: Exacerbations during pregnancy have been treated successfully with heme arginate or heme hydroxide (hematin); while neither preparation has been studied extensively during pregnancy, experience over many years suggests that treatment during pregnancy is unlikely to produce adverse fetal effects.
Genetic counseling.
VP is inherited in an autosomal dominant manner with reduced penetrance. De novo pathogenic variants are rare. Each child of an individual with VP has a 50% chance of inheriting the pathogenic variant; while offspring who inherit the variant may or may not develop manifestations, most do not. Prenatal testing for pregnancies at increased risk for VP is possible if the pathogenic variant in an affected family member has been identified. Of note, the presence of a PPOX pathogenic variant does not predict whether – or at what age – an individual will become symptomatic.
Diagnosis
Suggestive Findings
Variegate porphyria (VP) should be suspected in individuals with the following clinical findings and initial laboratory findings.
Clinical findings
- Cutaneous manifestations include chronic blistering photosensitivity, most commonly on the backs of the hands. Chronic features include blisters, milia, scarring, thickening, and areas of decreased and increased skin pigmentation. Facial hyperpigmentation and hypertrichosis may occur. The skin lesions are identical to those of porphyria cutanea tarda (PCT) and other blistering cutaneous porphyrias [Meissner et al 2003] (see Differential Diagnosis).
- Neurovisceral symptoms most commonly include the following:
- Abdominal pain. The pain is typically severe, steady rather than cramping, and diffuse rather than localized. Because the pain is neuropathic rather than inflammatory, abdominal findings are minimal compared to the severity of the pain. Ileus and bladder distension may be present. Acute hepatic porphyrias should be suspected whenever abdominal pain remains unexplained after an initial workup for common causes.
- Constipation
- Pain in the back, chest, and extremities
- Anxiety
- Seizures
- Muscle weakness due to a primarily motor neuropathy that usually begins in the proximal upper extremities and may progress to quadriparesis and respiratory paralysis. This is accompanied by pain and sometimes sensory loss. Hyperreflexia may be seen initially, followed by hyporeflexia as motor neuropathy progresses.
- Hyponatremia, which increases the risk for seizures. It may be a manifestation of hypothalamic involvement and the syndrome of inappropriate antidiuretic hormone secretion [Anderson et al 2005].
Initial biochemical laboratory findings. As VP may present with blistering cutaneous lesions on sun-exposed skin, neurovisceral symptoms or both, initial first-line testing aims to detect all porphyrias that can cause either skin or neurovisceral manifestations (see Differential Diagnosis).
- Blistering cutaneous porphyrias (including VP). When VP or any other blistering cutaneous porphyria is suspected, the recommended initial test is measurement of plasma or urine porphyrins. If elevated, further testing is needed to determine the type of porphyria or whether the porphyrin elevation – particularly in urine – represents nonspecific porphyrinuria rather than porphyria.
- Acute porphyrias (including VP). Measurement of urinary porphobilinogen (PBG)* and total porphyrins. Urine δ-aminolevulinic acid (ALA) is often measured at the same time as PBG but this is not necessary for initial screening.*Note: (1) If an acute porphyria is confirmed by substantial elevation of urinary PBG, treatment can be started, if appropriate, for symptoms of an acute attack (see Management, Treatment of Manifestations) while further biochemical testing is being performed to determine the type of acute porphyria (see Differential Diagnosis). (2) If PBG is normal, total porphyrins and ALA should be measured in the same urine sample, because total porphyrins often remain elevated longer than PBG. In ALA dehydratase-deficiency porphyria (ADP), the rarest type of porphyria, ALA and total porphyrins (but not PBG) are markedly elevated [Anderson et al 2005].
- Substantial elevation in erythrocyte porphyrins is not consistent with VP, and points to an erythropoietic porphyria as a cause of blistering skin manifestations and elevation of urine and plasma porphyrins. Alternatively, substantial erythrocyte protoporphyrin in an individual with VP may suggest a concurrent condition that elevates zinc protoporphyrin, such as iron deficiency, lead poisoning, or another erythrocyte disorder.
Establishing the Diagnosis
Biochemical Diagnosis
When initial biochemical laboratory findings support an acute porphyria (i.e., elevated urine PBG or porphyrins) or a blistering cutaneous porphyria (i.e., elevated plasma or urine porphyrins), further diagnostic biochemical testing (Table 1) is required to differentiate VP from other acute and cutaneous porphyrias and from conditions (e.g., liver disease) that cause nonspecific porphyrinuria:
- Plasma fluorescence scanning can establish or exclude VP when urine PBG is elevated since a fluorescence peak at ~626 nm is not found in any other type of porphyria.
- Fecal porphyrin analysis can differentiate VP, acute intermittent porphyria (AIP), and hereditary coproporphyria (HCP), the only diseases that substantially elevate urine PBG.
- Fecal porphyrin analysis and plasma fluorescence scanning can also reliably distinguish VP from porphyria cutanea tarda (PCT) and other porphyrias that cause blistering skin lesions (see Differential Diagnosis).
Table 1.
Biochemical Characteristics of Variegate Porphyria (VP)
Molecular Diagnosis
Identification of the causative pathogenic variant is now considered standard of care in VP and other acute porphyrias to confirm the diagnosis and inform genetic counseling (see Genetic Counseling) (see Option 1 and Option 2).
Option 1. It is generally preferred to establish the biochemical diagnosis of VP first, followed by confirmatory single-gene (PPOX) testing. However, a multigene panel (HMBS, CPOX, PPOX) can be used to establish the diagnosis when biochemical testing (e.g., substantial PBG elevation) indicates a diagnosis of AIP, HCP, or VP or when the individual to be tested has become asymptomatic and biochemical abnormalities are absent or nonspecific.
- Single-gene testing of PPOX is generally recommended after a diagnosis of VP is established biochemically. Sequence analysis detects small intragenic deletions/insertions and missense, nonsense, and splice site variants; typically, exon or whole-gene deletions/duplications are not detected. Standard practice is to perform sequence analysis first. If no PPOX pathogenic variant is found in an individual with biochemically proven VP, perform gene-targeted deletion/duplication analysis to detect intragenic deletions or duplications.Note: When VP is established biochemically in members of the Afrikaner population of South Africa, it is reasonable to consider targeted analysis for the founder variant, p.Arg59Trp, observed in about 95% of persons with variegate porphyria in that population [Dean 1971, Meissner et al 1996]. See Table 4. Notable PPOX Pathogenic Variants.
- An acute porphyria multigene panel that includes PPOX and other genes of interest (particularly HMBS and CPOX; see Differential Diagnosis) is most likely to identify the genetic cause of symptoms and PBG elevation while limiting identification of variants of uncertain significance and pathogenic variants in genes that do not explain the underlying phenotype.ALAD, the gene encoding the enzyme ALA dehydratase (which is deficient in ALA dehydratase-deficiency porphyria) may also be included in the panel, but is only relevant when ALA and porphyrins (but not PBG) are elevated.Note: (1) The diagnostic sensitivity of the testing used for each gene may vary by laboratory and is likely to change over time. (2) Methods used in a panel may include sequence analysis, deletion/duplication analysis, and/or other non-sequencing-based tests.
Option 2. Genomic testing is not recommended for initial diagnosis of acute porphyrias. However, when genomic testing obtained as part of a search for the cause of unexplained symptoms identifies HMBS, CPOX, or PPOX pathogenic variants or variants of uncertain significance, biochemical testing that documents elevations in PBG and porphyrins confirms porphyria as the cause of the symptoms – and confirms the diagnosis of VP.
For an introduction to comprehensive genomic testing click here. More detailed information for clinicians ordering genomic testing can be found here.
Table 2.
Molecular Genetic Testing Used in Variegate Porphyria
Clinical Characteristics
Clinical Description
Variegate porphyria (VP) is classified as both a cutaneous and an acute porphyria. It can present with chronic blistering cutaneous manifestations and/or acute attacks of neurovisceral manifestations that may become chronic.
Cutaneous manifestations. Chronic blistering photosensitivity, typically on the backs of the hands, is the most common manifestation of VP. The lesions result from sun exposure that activates porphyrins and makes the skin fragile and prone to blister formation. Lesions are located on sun-exposed areas, especially the dorsal aspects of the hands and less frequently the face, neck, ears, and lower extremities. Because sun-induced damage is not acute, the role of sunlight is often not recognized. Cutaneous manifestations may improve in winter and be less prevalent in northern regions and in dark-skinned individuals.
These and other manifestations of VP appear typically in adulthood and rarely before puberty.
The subepidermal vesicles, bullae, and erosions crust over and heal slowly. When blisters rupture they may become infected and painful.
Other chronic skin findings include milia, scarring, thickening, and areas of decreased and increased skin pigmentation. Facial hyperpigmentation and hypertrichosis may occur.
The skin manifestations are identical to those seen in porphyria cutanea tarda (PCT) and hereditary coproporphyria (HCP), and less severe than those seen in congenital erythropoietic porphyria (CEP) and hepatoerythropoietic porphyria (HEP). They contrast with the acute non-blistering photocutaneous manifestations of erythropoietic protoporphyria (EPP) (see Table 3).
Of note, the great majority of individuals who are heterozygous for a PPOX pathogenic variant are asymptomatic and are unlikely to be recognized unless they are screened for VP based on a family history of VP (see Genetic Counseling). In South Africa the frequency of acute attacks has decreased in recent decades. This may be due to less common use of harmful drugs such as barbiturates and sulfonamide antibiotics in clinical practice and perhaps better case recognition and better dissemination of information on how to avoid future attacks. VP now more commonly presents in South Africa with cutaneous rather than acute manifestations [Meissner et al 2003, Anderson et al 2005, Hift & Meissner 2005].
Neurovisceral symptoms can occur at any age after puberty as acute attacks, but may become chronic. Symptoms are more common in women than men, and occur less often in the elderly. The frequency and severity of attacks vary considerably and are determined, in part, by exacerbating factors such as certain drugs, hormones, and nutritional deficits [Anderson et al 2005]. The proportion of persons heterozygous for a PPOX pathogenic variant who experience acute attacks decreased from about 30%-40% in the 1980s to 5%-10% in 2005 [Hift & Meissner 2005].
The neurovisceral symptoms are identical to those in the other acute porphyrias (see Differential Diagnosis).
Acute manifestations vary. The most common symptoms are abdominal pain; nausea and vomiting; constipation; pain in the back, chest, and extremities; anxiety; seizures; and a predominantly motor peripheral neuropathy resulting in muscle weakness that may progress to quadriparesis and respiratory paralysis [Kauppinen & Mustajoki 1992, Meissner et al 2003, Anderson et al 2005, Hift & Meissner 2005]. Psychiatric disturbances and autonomic neuropathy can also be observed. Not all symptoms are present in a single episode and symptoms can vary from episode to episode; however, recurrent attacks are often similar. Acute attacks may be severe and are potentially fatal, but on average are less frequent and less severe than those observed in acute intermittent porphyria (AIP) [Hift & Meissner 2005].
Motor neuropathy usually manifests initially as proximal upper-extremity muscle weakness and can be difficult to detect. Hyperreflexia may be seen initially, followed by hyporeflexia as the motor neuropathy progresses. The motor neuropathy may be accompanied by sensory loss. Note: Motor neuropathy due to acute porphyrias is accompanied by little or no elevation of cerebrospinal fluid protein, which helps to differentiate it from the Landry Guillain-Barré syndrome [Anderson et al 2005].
Because abdominal pain is neuropathic rather than inflammatory, abdominal findings are minimal compared to the severity of the pain. Ileus and bladder distension may be present.
An acute attack can be fatal in the presence of severe manifestations including neuropathy, seizures, and respiratory compromise. If managed properly, the outcome of an acute attack is generally good. Even severe motor neuropathy is reversible with recovery over a variable period of months and sometimes over several years.
Factors that predispose to acute attacks that are often identified include exposure to a harmful drug, alcohol, reduced dietary intake, or stress from an infection or other illness. Most harmful drugs are known to be inducers of hepatic δ-aminolevulinic acid synthase (ALAS) and hepatic cytochrome P450 enzymes (see Agents/Circumstances to Avoid). Pregnancy is usually well tolerated but can precipitate acute attacks in some women.
Physical findings such as tachycardia, hypertension, restlessness, and agitation result from autonomic neuropathy and increased circulating catecholamines.
Chronic pain may be a manifestation of VP and other acute porphyrias. Depression may be more difficult to link to the disease. Chronic pain and depression may become important management issues.
Chronic liver abnormalities, particularly mild elevation of serum transaminases, are common. Risks for development of hepatocellular carcinoma and chronic renal disease are increased in VP (as well as in AIP and HCP). Hepatocellular carcinoma may develop, especially after age 50 years in persons with persistent elevations in porphobilinogen and porphyrins.
Note: The speculation that King George III (and perhaps others in the British royal family) had VP has been discounted [Peters 2011].
Genotype-Phenotype Correlations
PPOX pathogenic variants are generally severe and result in little or no enzyme activity; the residual approximately half-normal enzyme activity is a product of the normal allele. Therefore, different pathogenic variants are not associated with differences in disease severity [Whatley et al 1999, Whatley et al 2009].
Double heterozygosity for pathogenic variants in two different genes in the heme biosynthetic pathway. A patient with cutaneous manifestations initially diagnosed as HCP was found to be a double heterozygote for pathogenic variants in PPOX and CPOX after other family members were found to have clinical and biochemical features of VP [van Tuyll van Serooskerken et al 2011]. The phenotypes of such rare double heterozygotes are not necessarily more severe than the phenotype associated with heterozygosity for a pathogenic variant in one gene alone, suggesting that individuals who are doubly heterozygous for pathogenic variants in genes causing two different types of acute porphyria may be more common than has been assumed.
Note: Typically double heterozygosity is suspected because of unusual biochemical patterns, and thus is unlikely to be recognized without comprehensive biochemical testing [van Tuyll van Serooskerken et al 2011], which then identifies a need for additional molecular genetic testing.
Penetrance
PPOX pathogenic variants that result in VP produce little or no functional enzyme; the approximately 50% of normal residual enzyme activity results primarily from the normal allele. Penetrance is low, but may be increased by factors that increase the demand for hepatic heme synthesis. Penetrance is likely influenced by modifying genes that remain to be identified.
Nomenclature
Variegate porphyria (VP) and hereditary coproporphyria (HCP) were sometimes referred to as mixed porphyria, which is now an obsolete term.
VP has also been referred to as South African acute porphyria or protocoproporphyria.
In the past, familial porphyria cutanea tarda (PCT) may not have been clearly differentiated from VP in some instances.
Prevalence
It is estimated that in the South African population three individuals per 1,000 are heterozygous for the PPOX pathogenic variant p.Arg59Trp [Meissner et al 1996, Meissner et al 2003].
The prevalence of VP with present or past symptoms in Europe is about half that for acute intermittent porphyria (AIP), and has been estimated at 3.2:1,000,000 [Elder et al 2013].
Genetically Related (Allelic) Disorders
Individuals with biallelic PPOX pathogenic variants (i.e., homozygotes or compound heterozygotes) have been described on rare occasions [Frank et al 1998, Pinder et al 2013]. In childhood they manifest neurologic findings including intellectual disability and/or seizures and cutaneous manifestations only. In these individuals one or both alleles must produce some PPOX enzyme.
Differential Diagnosis
The genetic porphyrias comprise a group of distinct diseases, each resulting from alteration of a specific step in the heme synthesis pathway that results in characteristic patterns of accumulation of pathway intermediates (Figure 1).
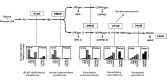
In Table 3 the porphyrias are grouped by their principal clinical manifestations (neurovisceral or cutaneous) and the tissue origin of the excess production of pathway intermediates: liver (i.e., hepatic); or bone marrow (i.e., erythropoietic).
Porphyrias with neurologic manifestations are considered acute because the symptoms usually occur as discrete, severe episodes, which may be induced by endogenous hormones, drugs and dietary changes; they are difficult to diagnose due to their rarity and the nonspecific nature of symptoms, even when severe. The four acute porphyrias (often referred to as acute hepatic porphyrias) are: ALA dehydratase deficiency porphyria (ADP), acute intermittent porphyria (AIP), hereditary coproporphyria (HCP), and variegate porphyria (VP). Only a few individuals with ADP have been reported in the world literature, and whether this porphyria is hepatic, erythropoietic, or both is uncertain.
Porphyrias with cutaneous manifestations include those causing chronic blistering skin lesions (i.e., VP as well as porphyria cutanea tarda [PCT], HCP, congenital erythropoietic porphyria [CEP], and hepatoerythropoietic porphyria [HEP]) or acute non-blistering photosensitivity (i.e., EPP and XLP).
Table 3.
Classification of the Hereditary Porphyrias
Acute neurologic porphyrias. The acute neurovisceral symptoms of VP are identical to those of the other acute porphyrias. VP can be differentiated from AIP and HCP by plasma fluorescence scanning and fecal porphyrin analysis (Table 1) or by molecular genetic testing (Table 2).
In individuals with progressive weakness due to the motor neuropathy caused by one of the acute porphyrias (AIP, VP, HCP, and ADP), the entity most likely to be considered is acute ascending polyneuropathy, the Landry Guillain-Barré syndrome.
- Abdominal pain, constipation, and tachycardia usually accompany the acute neurologic illness in the acute porphyrias but not in Landry Guillain-Barré syndrome.
- CSF protein is usually normal in the acute porphyrias, but usually elevated in Landry Guillain-Barré syndrome.
- Most importantly, urinary PBG is markedly elevated in the acute porphyrias especially when symptoms are present, but normal in Landry Guillain-Barré syndrome.
Chronic blistering photocutaneous porphyrias. VP can be readily differentiated from the following conditions by biochemical testing and ultimately confirmed by molecular genetic testing.
- The blistering skin lesions of porphyria cutanea tarda (PCT), the most common human porphyria, are identical to those of VP. Because PCT is much more common than VP, patients with VP are often misdiagnosed as having PCT. Because treatment for PCT is not effective in VP, it is important to differentiate these disorders before initiating treatment.
- HCP is associated with such skin manifestations much less commonly than VP.
- Blistering skin manifestations occur in AIP only when concurrent end-stage renal disease impairs porphyrin excretion, and thus increases plasma porphyrin levels.
- Cutaneous manifestations of CEP and HEP are also chronic and blistering but usually more severe than those of VP because circulating porphyrin levels are usually much higher (often by an order of magnitude) than in PCT and VP. Although the diagnosis in individuals with mild CEP and HEP is readily mistaken for VP, HCP, and PCT during clinical evaluation, these erythropoietic porphyrias are differentiated particularly by finding high levels of erythrocyte porphyrins.
- Pseudoporphyria is a little-understood condition with cutaneous findings similar to PCT and VP but without significant porphyrin elevations.
Management
Evaluations Following Initial Diagnosis
To establish the extent of disease and to plan the management of an individual diagnosed with variegate porphyria (VP), the following clinical and laboratory evaluations (if not performed as part of the evaluation that led to the diagnosis) are recommended:
- Degree of elevations on plasma and urine porphyrins and urine porphobilinogen (PBG), if not determined at the time of diagnosis
- Clinical evaluation of any current acute neurovisceral manifestations to determine the need for hospital admission and treatment with hemin.
- Nervous system. Assessment of the extent of neurologic involvement causing paresis, pain or sensory changes
- Psychiatric evaluation if depression or other psychiatric features are present
- Liver. Liver function tests to indicate chronic liver involvement and liver imaging in patients older than age 50 years
- Kidneys. Kidney function tests to assess for presence and progression of kidney damage
- Skin. Assessment of blistering cutaneous lesions to assess their relationship to VP
- Contributions of medications (see Agents/Circumstances to Avoid), diet, and concurrent conditions to severity of VP
- Consultation with a clinical geneticist and/or genetic counselor
Treatment of Manifestations
Neurovisceral Symptoms
Most acute neurovisceral attacks require hospital admission; patients with mild attacks (not requiring narcotic analgesics and without hyponatremia, seizures, or muscle weakness) are sometimes treated as outpatients. A rapid, thorough, and multidisciplinary evaluation is optimized by in-patient management.
As with other acute porphyrias, evaluation should include identification of exacerbating drugs and other precipitating factors. Harmful medications include barbiturates, sulfonamide antibiotics, griseofulvin, rifampin, most anticonvulsants including phenytoin and carbamazepine, alcohol, ergot alkaloids, metoclopramide, and progestins. Harmful medications should be discontinued [Balwani et al 2017].
Seizures, motor neuropathy, and hyponatremia suggest severe disease and should be managed in the ICU with adequate supportive treatment. Evidence of reversible cerebral vasospasm may be found by MRI [Webb et al 2016].
Narcotic analgesics are usually required for pain and ondansetron or a related drug for nausea and vomiting. A phenothiazine is also effective for nausea and for psychiatric symptoms (e.g., agitation, hallucinations) [Anderson et al 2005, Harper & Wahlin 2007].
Mild attacks (not requiring narcotics and without hyponatremia, seizures, or motor neuropathy) can be treated with glucose loading, but most attacks should be treated with intravenous hemin [Anderson et al 2005, Balwani et al 2017].
Note: "Hemin" refers to the oxidized form of iron protoporphyrin IX, but is also the generic term for heme preparations used as intravenous therapies for acute porphyrias, such as lyophilized hematin (heme hydroxide) and heme arginate. When these hemin preparations are infused intravenously, the heme is bound to circulating albumin as heme albumin. The latter is taken up by hepatocytes and decreases the synthesis of hepatic ALAS1, the rate-controlling enzyme for heme synthesis in the liver.
Patients with acute attack should be carefully monitored for muscle weakness and respiratory impairment that may require ventilatory support.
Hyponatremia should be corrected slowly and seizures treated with medications that do not exacerbate porphyria.
Liver transplantation, which has been effective in persons with acute intermittent porphyria with severe repeated acute attacks that respond poorly to medical therapy, is also a consideration in VP [Dowman et al 2012].
Progression of renal disease may be prevented to some degree by controlling hypertension.
Cutaneous Manifestations
Porphyrin levels may decrease and photosensitivity improve if exacerbating factors can be identified and removed; otherwise, there is no effective treatment that lowers porphyrin levels. Treatment with hemin may lower porphyrins in the short term only.
Patients should wear protective clothing and avoid exposure to sunlight.
Analgesics may be needed for painful lesions and antibiotics for superimposed infection. Topical steroids are of little or no benefit.
Specific measures effective in the treatment of porphyria cutanea tarda (i.e., phlebotomy and low-dose hydroxychloroquine or chloroquine) are not effective in the management of VP.
Prevention of Primary Manifestations
Acute attack
- Attacks are less likely to occur in the future if exacerbating factors are corrected or avoided (see Agents/Circumstances to Avoid).
- Recurrent premenstrual attacks of acute porphyrias, including VP, can be prevented with gonadotropin-releasing hormone analogs [Anderson et al 1990, Schulenburg-Brand et al 2017].
- Weekly or biweekly hemin infusions may prevent frequent noncyclical attacks; however, published experience is lacking [Marsden et al 2015].
- Givosiran, a small interfering RNA (siRNA) therapeutic, was recently approved by the FDA for treatment of acute porphyrias, including VP. In particular, monthly subcutaneous injections of givosiran can be effective for prevention of frequently recurring attacks [Sardh et al 2019].
Photocutaneous manifestations. Prevention of the skin manifestations of VP requires protection from sunlight. Avoidance of exacerbating factors may also be beneficial.
Surveillance
Hepatocellular carcinoma may develop especially after age 50 years in patients with acute porphyrias and persistent elevations in porphobilinogen or porphyrins; liver imaging at six-month intervals beginning at age 50 years may detect early lesions [Andant et al 2000, Schneider-Yin et al 2010].
Agents/Circumstances to Avoid
Precipitating factors that should be avoided include: barbiturates, sulfonamide antibiotics, griseofulvin, rifampin, most anticonvulsants including phenytoin and carbamazepine, alcohol, ergot alkaloids, metoclopramide, and progestins.
Updated lists are maintained at the websites of the American Porphyria Foundation and the European Porphyria Network.
Although birth control pills should generally be avoided, low-dose hormonal preparations may be tolerated.
Fasting and very low calorie diets should be avoided. Bariatric surgery should be avoided in patients who have had frequent exacerbations of VP and other acute porphyrias. Patients who wish to lose weight should do so gradually with moderate, long-term reductions in calorie intake under guidance of a dietician.
Evaluation of Relatives at Risk
It is appropriate to clarify the genetic status of apparently asymptomatic older and younger at-risk relatives of a proband. This will identify individuals heterozygous for the familial PPOX pathogenic variant who may benefit from counseling regarding appropriate use of drugs and avoidance of known precipitating factors.
See Genetic Counseling for issues related to testing of at-risk relatives for genetic counseling purposes.
Pregnancy Management
Pregnancy is usually well tolerated in women with variegate porphyria (VP); however, some women with VP may experience exacerbations during pregnancy.
Badminton & Deybach [2006] published an anecdotal report of successful treatment of several pregnant women experiencing attacks of VP or other acute porphyrias during pregnancy with hemin (in the form of heme arginate) without adverse fetal effect. They emphasize that interruption of pregnancy is almost never indicated for management of acute porphyria.
Experience with heme hydroxide (hematin) is also limited but suggests no adverse effects during pregnancy [Isenschmid et al 1992]. As noted (see Treatment of Manifestations, Neurovisceral Symptoms), hemin is delivered to tissues as heme albumin when administered as either heme arginate or heme hydroxide, and these preparations are expected to have similar safety profiles.
A fetus heterozygous for a PPOX pathogenic variant has a good prognosis, because current postnatal management involves counseling the family regarding appropriate use of drugs and avoidance of known precipitating factors.
See MotherToBaby for further information on medication use during pregnancy.
Therapies Under Investigation
Search ClinicalTrials.gov in the US and EU Clinical Trials Register in Europe for access to information on clinical studies for a wide range of diseases and conditions. Note: There may not be clinical trials for this disorder.
Genetic Counseling
Genetic counseling is the process of providing individuals and families with information on the nature, mode(s) of inheritance, and implications of genetic disorders to help them make informed medical and personal decisions. The following section deals with genetic risk assessment and the use of family history and genetic testing to clarify genetic status for family members; it is not meant to address all personal, cultural, or ethical issues that may arise or to substitute for consultation with a genetics professional. —ED.
Mode of Inheritance
Variegate porphyria (VP) is inherited in an autosomal dominant manner with reduced penetrance.
Risk to Family Members
Parents of a proband
- Typically, one parent of an individual diagnosed with VP is heterozygous for the PPOX pathogenic identified in the proband and may or may not have had symptoms.
- Rarely, an individual diagnosed with VP may have the disorder as the result of a de novo pathogenic variant.
- Molecular genetic testing is recommended for the parents of a proband with an apparent de novo pathogenic variant.
- If the pathogenic variant found in the proband cannot be detected in the leukocyte DNA of either parent, possible explanations include a de novo pathogenic variant in the proband or germline mosaicism in a parent. Though theoretically possible, no instances of a proband inheriting a pathogenic variant from a parent with germline mosaicism have been reported.
- The family history of some individuals diagnosed with VP may appear to be negative because of reduced penetrance or failure to recognize the disorder as the cause of nonspecific symptoms in family members. Therefore, an apparently negative family history cannot be confirmed unless molecular genetic testing has been performed on the parents of the proband.
Sibs of a proband. The risk to the sibs of the proband depends on the genetic status of the proband's parents:
- If a parent of the proband has the PPOX pathogenic variant identified in the proband, each sib has a 50% chance of inheriting the variant. A sib who inherits the pathogenic variant may or may not develop symptoms of VP.
- If the proband has a known VP-related pathogenic variant that cannot be detected in the leukocyte DNA of either parent, the recurrence risk to sibs is estimated to be 1% because of the theoretic possibility of parental germline mosaicism [Rahbari et al 2016].
Offspring of a proband
- Each child of an individual with VP has a 50% chance of inheriting the pathogenic variant.
- Offspring who inherit the pathogenic variant may or may not develop symptoms.
Other family members
- The risk to other family members depends on the status of the proband's parents: if a parent has the PPOX pathogenic variant, his or her family members may be at risk.
- A family member with the PPOX pathogenic variant may or may not develop symptoms of VP.
Related Genetic Counseling Issues
See Management, Evaluation of Relatives at Risk for information on evaluating at-risk relatives for the purpose of early diagnosis and treatment.
Considerations in families with an apparent de novo pathogenic variant. When neither parent of a proband with an autosomal dominant condition has the pathogenic variant identified in the proband or clinical evidence of the disorder, the pathogenic variant is likely de novo. However, non-medical explanations including alternate paternity or maternity (e.g., with assisted reproduction) and undisclosed adoption could also be explored.
Family planning
- The optimal time for determination of genetic risk and discussion of the availability of prenatal/preimplantation genetic testing is before pregnancy.
- It is appropriate to offer genetic counseling (including discussion of potential risks to offspring and reproductive options) to young adults who are affected or at risk.
DNA banking. Because it is likely that testing methodology and our understanding of genes, pathogenic mechanisms, and diseases will improve in the future, consideration should be given to banking DNA from probands in whom a molecular diagnosis has not been confirmed (i.e., the causative pathogenic mechanism is unknown).
Prenatal Testing and Preimplantation Genetic Testing
Once the PPOX pathogenic variant has been identified in an affected family member, prenatal testing for a pregnancy at increased risk and preimplantation genetic testing for VP are possible. Note: The presence of a PPOX pathogenic variant detected by prenatal testing does not predict whether individuals will be symptomatic, or if they are, the age of onset or presentation of the disorder.
Differences in perspective may exist among medical professionals and within families regarding the use of prenatal testing, particularly if the testing is being considered for the purpose of pregnancy termination rather than early diagnosis. While most centers would consider use of prenatal testing to be a personal decision, discussion of these issues may be helpful.
Resources
GeneReviews staff has selected the following disease-specific and/or umbrella support organizations and/or registries for the benefit of individuals with this disorder and their families. GeneReviews is not responsible for the information provided by other organizations. For information on selection criteria, click here.
- American Porphyria Foundation (APF)4915 St. Elmo AvenueSuite 200Bethesda MD 20814Phone: 1-866-APF-3635 (toll-free); 301-347-7166Email: [email protected]
- British Porphyria AssociationUnited KingdomPhone: 0300 30 200 30Email: [email protected]
- Canadian Association for Porphyria/Association Canadienne de PorphyrieCanada
- Find a Porphyria ExpertAmerican Porphyria Foundation
- MedlinePlus
- Porphyria South AfricaSouth AfricaPhone: +27 21-4066332Fax: +27 21-4066061Email: [email protected]
- Welsh Medicines Information CentreThe Welsh Medicines Information Centre (WMIC) offers a specialist advisory service on the safe use of drugs in porphyria.United KingdomPhone: +44 029 2074 4298
- Global Porphyria Advocacy Coalition
- International Porphyria NetworkEmail: [email protected]
- Swedish Porphyria AssociationSwedenPhone: +46730803820Email: [email protected]
Molecular Genetics
Information in the Molecular Genetics and OMIM tables may differ from that elsewhere in the GeneReview: tables may contain more recent information. —ED.
Table A.
Variegate Porphyria: Genes and Databases
Table B.
OMIM Entries for Variegate Porphyria (View All in OMIM)
Molecular Pathogenesis
PPOX catalyzes the oxidation of protoporphyrinogen to protoporphyrin, with removal of six protons. The partial deficiency of PPOX in VP limits heme synthesis, particularly in the presence of factors that lead to increased hepatic heme synthesis and induction of δ-aminolevulinic acid synthase-1 (ALAS1) in the liver. ALAS1 is the ubiquitous form of ALAS, which is found in all tissues, in contrast to the erythroid-specific form known as ALAS2, which is produced only in the bone marrow.
Mechanism of disease causation. VP is caused by heterozygous loss-of-function PPOX variants, resulting in reduction of the enzyme in all tissues to approximately 50% of normal.
Table 4.
Notable PPOX Pathogenic Variants
References
Literature Cited
- Andant C, Puy H, Bogard C, Faivre J, Soulé JC, Nordmann Y, Deybach JC. Hepatocellular carcinoma in patients with acute hepatic porphyria: frequency of occurrence and related factors. J Hepatol. 2000;32:933–9. [PubMed: 10898313]
- Anderson KE, Bloomer JR, Bonkovsky HL, Kushner JP, Pierach CA, Pimstone NR, Desnick RJ. Recommendations for the diagnosis and treatment of the acute porphyrias. Ann Intern Med. 2005;142:439–50. [PubMed: 15767622]
- Anderson KE, Spitz IM, Bardin CW, Kappas A. A gonadotropin releasing hormone analogue prevents cyclical attacks of porphyria. Arch Intern Med. 1990;150:1469–74. [PubMed: 2196028]
- Badminton MN, Deybach JC. Treatment of an acute attack of porphyria during pregnancy. Eur J Neurol. 2006;13:668–9. [PubMed: 16796597]
- Balwani M, Wang B, Anderson KE, Bloomer JR, Bissell DM, Bonkovsky HL, Phillips JD, Desnick RJ. Acute hepatic porphyrias: recommendations for evaluation and long-term management. Hepatology. 2017;66:1314–1322. [PMC free article: PMC5605422] [PubMed: 28605040]
- Barbaro M, Kotajärvi M, Harper P, Floderus Y. Partial protoporphyrinogen oxidase (PPOX) gene deletions, due to different Alu-mediated mechanisms, identified by MLPA analysis in patients with variegate porphyria. Orphanet J Rare Dis. 2013;8:13. [PMC free article: PMC3554555] [PubMed: 23324528]
- Dean G. The Porphyrias: A Study of Inheritance and Environment. 2 ed. London, UK: Pitman Medical; 1971.
- Dowman JK, Gunson BK, Mirza DF, Bramhall SR, Badminton MN, Newsome PN, et al. Liver transplantation for acute intermittent porphyria is complicated by a high rate of hepatic artery thrombosis. Liver Transpl. 2012;18:195–200. [PMC free article: PMC3472026] [PubMed: 21618697]
- Elder G, Pauline Harper P, Michael Badminton M, Sandberg S, Deybach J-C. The incidence of inherited porphyrias in Europe. J Inherit Metab Dis. 2013;36:849–57. [PubMed: 23114748]
- Frank J, McGrath J, Lam H, Graham RM, Hawk JL, Christiano AM. Homozygous variegate porphyria: identification of mutations on both alleles of the protoporphyrinogen oxidase gene in a severely affected proband. J Invest Dermatol. 1998;110:452–5. [PubMed: 9540991]
- Harper P, Wahlin S. Treatment options in acute porphyria, porphyria cutanea tarda, and erythropoietic protoporphyria. Curr Treat Options Gastroenterol. 2007;10:444–55. [PubMed: 18221605]
- Hift RJ, Meissner PN. An analysis of 112 acute porphyric attacks in Cape Town, South Africa: Evidence that acute intermittent porphyria and variegate porphyria differ in susceptibility and severity. Medicine (Baltimore). 2005;84:48–60. [PubMed: 15643299]
- Isenschmid M, König C, Fässli C, Haenel A, Hänggi W, Schneider H. Acute intermittent porphyria in pregnancy: glucose or hematin therapy? Schweiz Med Wochenschr. 1992;122:1741–5. [PubMed: 1448679]
- Kauppinen R, Mustajoki P. Prognosis of acute porphyria: occurrence of acute attacks, precipitating factors, and associated diseases. Medicine (Baltimore). 1992;71:1–13. [PubMed: 1549056]
- Marsden JT, Guppy S, Stein P, Cox TM, Badminton M, Gardiner T, Barth JH, Stewart MF, Rees DC. Audit of the use of regular haem arginate Infusions in patients with acute porphyria to prevent recurrent symptoms. JIMD Reports. 2015;22:57–65. [PMC free article: PMC4486272] [PubMed: 25762493]
- Mauzerall D, Granick S. The occurrence and determination of δ-aminolevulinic acid and porphobilinogen in urine. J Biol Chem. 1956;219:435–46. [PubMed: 13295297]
- Meissner P, Hift RJ, Corrigall A. Variegate porphyria. In: Kadish KM, Smith K, Guilard R, eds. Porphyrin Handbook, Part II. Vol 14. San Diego, CA: Academic Press; 2003:93-120.
- Meissner PN, Dailey TA, Hift RJ, Ziman M, Corrigall AV, Roberts AG, Meissner DM, Kirsch RE, Dailey HA. A. R59W mutation in human protoporphyrinogen oxidase results in decreased enzyme activity and is prevalent in South Africans with variegate porphyria. Nat Genet. 1996;13:95–7. [PubMed: 8673113]
- Peters T. King George III, bipolar disorder, porphyria and lessons for historians. Clin Med (Lond). 2011;11:261–4. [PMC free article: PMC4953321] [PubMed: 21902081]
- Pinder VA, Holden ST, Deshpande C, Siddiqui A, Mellerio JE, Wraige E, Powell AM. Homozygous variegate porphyria presenting with developmental and language delay in childhood. Clin Exp Dermatol. 2013;38:737–40. [PubMed: 24073655]
- Poh-Fitzpatrick MB. A plasma porphyrin fluorescence marker for variegate porphyria. Arch Dermatol. 1980;116:543–7. [PubMed: 7377785]
- Rahbari R, Wuster A, Lindsay SJ, Hardwick RJ, Alexandrov LB, Turki SA, Dominiczak A, Morris A, Porteous D, Smith B, Stratton MR, Hurles ME, et al. Timing, rates and spectra of human germline mutation. Nat Genet. 2016;48:126–33. [PMC free article: PMC4731925] [PubMed: 26656846]
- Sardh E, Harper P, Balwani B, Stein P, Rees D, Bissell DM, Desnick R, Parker C, Phillips J, Bonkovsky HL, Vassiliou D, Penz C, Chan-Daniels A, He Q, Querbes W, Fitzgerald K, Kim JB. MD, Garg P, Vaishnaw A, Simon AR, Anderson KE, A phase 1 study of RNA interference therapy for acute intermittent porphyria. NEJM. 2019;380:549–58. [PubMed: 30726693]
- Schneider-Yin X, van Tuyll van Serooskerken AM, Went P, Tyblewski W, Poblete-Gutiérrez P, Minder EI, Frank J. Hepatocellular carcinoma in variegate porphyria: a serious complication. Acta Derm Venereol. 2010;90:512–5. [PubMed: 20814629]
- Schulenburg-Brand D, Gardiner T, Guppy S, Rees DC, Stein P, Barth J, Stewart MF, Badminton M. An audit of the use of gonadorelin analogues to prevent recurrent acute symptoms in patients with acute porphyria in the United Kingdom. JIMD Reports. 2017;36:99–107. [PMC free article: PMC5680288] [PubMed: 28220410]
- van Tuyll van Serooskerken AM, de Rooij FW, Edixhoven A, Bladergroen RS, Baron JM, Joussen S, Merk HF, Steijlen PM, Poblete-Gutiérrez P, te Velde K, Wilson JH, Koole RH, van Geel M, Frank J. Digenic inheritance of mutations in the coproporphyrinogen oxidase and protoporphyrinogen oxidase genes in a unique type of porphyria. J Invest Dermatol. 2011;131:2249–54. [PubMed: 21734717]
- Webb AJS, Ingale H, Irani SR, Hu MTM. Acute variegate porphyria presenting with reversible cerebral vasoconstriction. Clin Neurol Neurosurg. 2016;146:102–4. [PubMed: 27186968]
- Whatley SD, Mason NG, Woolf JR, Newcombe RG, Elder GH, Badminton MN. Diagnostic strategies for autosomal dominant acute porphyrias: retrospective analysis of 467 unrelated patients referred for mutational analysis of the HMBS, CPOX, or PPOX gene. Clin Chem. 2009;55:1406–14. [PubMed: 19460837]
- Whatley SD, Puy H, Morgan RR, Robreau AM, Roberts AG, Nordmann Y, Elder GH, Deybach JC. Variegate porphyria in Western Europe: identification of PPOX gene mutations in 104 families, extent of allelic heterogeneity, and absence of correlation between phenotype and type of mutation. Am J Hum Genet. 1999;65:984–94. [PMC free article: PMC1288269] [PubMed: 10486317]
Chapter Notes
Acknowledgments
This work is supported by the Porphyrias Consortium funded by the NIH/NIDDK and part of the Rare Diseases Clinical Research Network, and the American Porphyria Foundation.
Revision History
- 12 December 2019 (bp) Comprehensive update posted live
- 14 February 2013 (me) Review posted live
- 12 June 2012 (kea) Original submission
Publication Details
Author Information and Affiliations
Publication History
Initial Posting: February 14, 2013; Last Update: December 12, 2019.
Copyright
GeneReviews® chapters are owned by the University of Washington. Permission is hereby granted to reproduce, distribute, and translate copies of content materials for noncommercial research purposes only, provided that (i) credit for source (http://www.genereviews.org/) and copyright (© 1993-2024 University of Washington) are included with each copy; (ii) a link to the original material is provided whenever the material is published elsewhere on the Web; and (iii) reproducers, distributors, and/or translators comply with the GeneReviews® Copyright Notice and Usage Disclaimer. No further modifications are allowed. For clarity, excerpts of GeneReviews chapters for use in lab reports and clinic notes are a permitted use.
For more information, see the GeneReviews® Copyright Notice and Usage Disclaimer.
For questions regarding permissions or whether a specified use is allowed, contact: ude.wu@tssamda.
Publisher
University of Washington, Seattle, Seattle (WA)
NLM Citation
Singal AK, Anderson KE. Variegate Porphyria. 2013 Feb 14 [Updated 2019 Dec 12]. In: Adam MP, Feldman J, Mirzaa GM, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2024.
