Summary
Clinical characteristics.
Nonketotic hyperglycinemia (NKH) is the inborn error of glycine metabolism defined by deficient activity of the glycine cleavage enzyme system (GCS), which results in accumulation of large quantities of glycine in all body tissues including the brain. Based on ultimate outcome NKH is categorized into severe NKH (no developmental progress and intractable epilepsy) and attenuated NKH (variable developmental progress and treatable or no epilepsy). The majority of children with NKH have onset in the neonatal period manifest as progressive lethargy evolving into profound coma and marked hypotonia; 85% have severe NKH and 15% attenuated NKH. Those with onset between two weeks and three months typically present with hypotonia; 50% have severe NKH and 50% attenuated NKH. Those with onset after age three months have attenuated NKH. Severe versus attenuated NKH is consistent within families, but the degree of developmental progress in those with attenuated NKH can vary.
Diagnosis/testing.
The diagnosis of NKH is established in a proband with elevated glycine in plasma and CSF, a compatible pattern on brain imaging, and either biallelic pathogenic variants in one of the genes encoding the protein subunits of the GCS identified on molecular genetic testing or deficient activity of the GCS (without deficiency of cofactors such as enzyme-bound lipoate or pyridoxal phosphate).
Management.
Treatment of manifestations:
- Severe NKH. No treatment is effective in changing the natural history of developmental delays, spasticity, and intractable epilepsy, but treatment with benzoate to lower glycine improves attentiveness and facilitates seizure management.
- Attenuated NKH. Current treatment is reduction of plasma concentration of glycine by administration of sodium benzoate and blockade of overstimulated NMDA receptors.
Surveillance: In the first years of life: routine developmental assessments and neurologic evaluations. Monitoring for scoliosis and hip dysplasia in severely affected individuals; gastrointestinal issues; and pulmonary function particularly in children who develop recurrent respiratory infections.
Agents/circumstances to avoid: Valproate, which raises blood and CSF glycine concentrations and may increase seizure frequency; vigabatrin, which has resulted in rapid loss of function when used to treat seizures, particularly in those with attenuated NKH who have West syndrome.
Genetic counseling.
NKH is inherited in an autosomal recessive manner. The parents of an affected individual are typically heterozygotes (i.e., carriers of one NKH-related pathogenic variant); however, de novo pathogenic variants occur in approximately 1% of individuals with NKH. If both parents are heterozygous for one pathogenic variant, each sib of an affected individual has at conception a 25% chance of being affected, a 50% chance of being an asymptomatic carrier, and a 25% chance of being unaffected and not a carrier. Once the pathogenic variants in an NKH-related gene have been identified in an affected family member, carrier testing for at-risk relatives and prenatal and preimplantation genetic testing are possible.
GeneReview Scope
Table
Severe NKH Attenuated NKH
Diagnosis
Suggestive Findings
Nonketotic hyperglycinemia (NKH) due to biallelic pathogenic variants in one of the two genes (GLDC and AMT) known to encode the components of the glycine cleavage enzyme system or possibly in a third gene (GCSH) should be suspected in individuals with the following clinical, laboratory, and neuroimaging findings.
Clinical findings
- Neonates with hypotonia, lethargy, coma, apnea, seizures with or without a burst suppression pattern on EEG
- Infants with lethargy, hypotonia, seizures, poor feeding, developmental delays
- Children with developmental delays (with expressive language more impaired than receptive language), hyperactivity with or without choreatic movements, particularly with episodic worsening of manifestations
- Individuals with isolated elevated levels of plasma glycine, particularly when associated with hyperactivity, developmental delays, and/or seizures, or any of the other above manifestations
Laboratory findings
- The combination of isolated elevation of levels of glycine in plasma and CSF (obtained simultaneously) by quantitative amino acid analysis (Table 1) and an abnormal CSF-to-plasma glycine ratio makes the likelihood of NKH high and requires confirmatory testing (see Establishing the Diagnosis).Note: (1) Accurate measurement of CSF glycine requires that the CSF be completely free of contamination by blood or serum (which is not visible to the eye), as evidenced by a normal RBC (red blood cell) count and protein concentration. The presence of blood or elevated protein in the CSF invalidates the results. (2) The elevation of CSF glycine is more important than the ratio, which is only a secondary measure. (3) In CSF, the serine concentration can be low, but the threonine concentration should not be elevated. (4) The elevation of glycine levels in CSF in NKH is usually higher than that observed in disorders affecting the cofactors of the glycine cleavage enzyme system (lipoate, pyridoxal phosphate) and overlaps with attenuated NKH, but exceptions exist [Mills et al 2010, Baker et al 2014]. (5) Documentation of a normal level of pyridoxal phosphate in the CSF helps to exclude disorders of pyridoxal phosphate metabolism, which can similarly raise CSF glycine levels. Further testing is needed to distinguish between the various glycine encephalopathies (see Differential Diagnosis).
- Urine organic acid profile is expected to be normal. Small elevations of multiple acylglycine esters can occasionally be noticed.
Table 1.
CSF and Plasma Glycine Concentration (µmol/L) in Nonketotic Hyperglycinemia (NKH)
Brain MRI
- The most consistent abnormalities are noted on diffusion-weighted imaging in the first three months of life, when the vast majority of individuals with NKH present clinically. All infants with NKH have diffusion restriction in the posterior limb of the internal capsule, anterior brain stem, posterior tegmental tracts, and cerebellum (see Figure 1) [Stence et al 2019].
- While the diffusion restriction in the infratentorial regions recedes after age three months, it often extends upwards to the motor cortex and a generalized diffusion restriction of the supratentorial white matter can be recognized between ages three and 14 months.
- Other
- The corpus callosum can be thin and shortened but is not absent.
- A small group of infants develop hydrocephalus, often with an enlarged retrocerebellar cystic region.
- Atrophy is present in older individuals with severe NKH, but often not in individuals with attenuated NKH.
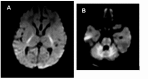
Figure 1.
Diffusion-weighted images of a neonate with classic NKH showing diffusion restriction: A. At the level of the posterior limb of the internal capsule; and
Brain magnetic resonance spectroscopy (MRS). On short echo time (TE = 35 msec) MRS, the glycine signal at 3.55 ppm coincides with myoinositol; however, at intermediate echo time (TE = 135 msec), glycine is recognized at 3.6 ppm without overlap. In most affected individuals with severe NKH a clear glycine peak is present, whereas in attenuated NKH the glycine peak is lower and sometimes difficult to detect [Heindel et al 1993, Gabis et al 2001, Stence et al 2019].
Establishing the Diagnosis
The diagnosis of NKH is established in a proband with elevated glycine in plasma and CSF (Table 1), a compatible pattern on brain imaging, and either biallelic pathogenic (or likely pathogenic) variants in one of the genes encoding the protein subunits of the GCS identified on molecular genetic testing (Table 2) or deficient activity of the GCS (without deficiency of cofactors such as enzyme-bound lipoate or pyridoxal phosphate). Today, confirmatory testing is primarily by molecular genetic testing; enzymatic testing is used only in select cases.
Note: (1) Per ACMG/AMP variant interpretation guidelines, the terms "pathogenic variant" and "likely pathogenic variant" are synonymous in a clinical setting, meaning that both are considered diagnostic and can be used for clinical decision making [Richards et al 2015]. Reference to "pathogenic variants" in this GeneReview is understood to include likely pathogenic variants. (2) The identification of variant(s) of uncertain significance cannot be used to confirm or rule out the diagnosis.
Molecular genetic testing. GLDC (encoding the GCS P-protein component) and AMT (encoding the GCS T-protein component) are the two genes in which biallelic pathogenic variants are known to cause NKH. Biallelic pathogenic variants in GCSH (encoding the GCS H-protein component) have been proposed as a cause of NKH in two individuals [C Acquaviva, P Rodríguez-Pomb, personal communications]; however, this remains unconfirmed.
Currently the most common testing strategy is to perform concurrent testing of all three genes (GLDC, AMT, and GCSH) by use of a multigene panel that includes these three genes and other genes of interest (see Differential Diagnosis). For this disorder a multigene panel that also includes deletion/duplication analysis is recommended (see Table 1).
The following considerations regarding multigene panels are offered by GeneReviews: Such a multigene panel is most likely to identify the genetic cause of the condition while limiting identification of variants of uncertain significance and pathogenic variants in genes that do not explain the underlying phenotype. Note: (1) The genes included in the panel and the diagnostic sensitivity of the testing used for each gene vary by laboratory and are likely to change over time. (2) Some multigene panels may include genes not associated with the condition discussed in this GeneReview. (3) In some laboratories, panel options may include a custom laboratory-designed panel and/or custom phenotype-focused exome analysis that includes genes specified by the clinician. (4) Methods used in a panel may include sequence analysis, deletion/duplication analysis, and/or other non-sequencing-based tests.
For an introduction to multigene panels click here. More detailed information for clinicians ordering genetic tests can be found here.
Table 2.
Molecular Genetic Testing Used in Nonketotic Hyperglycinemia (NKH)
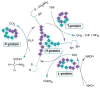
Analysis of the activity of the glycine cleavage enzyme system (GCS), the major degradative pathway for glycine (see Figure 2), requires analysis of a liver biopsy, usually obtained by surgical endoscopy as a wedge biopsy or as soon as possible at autopsy. Because the enzyme is labile, rapid processing and deep freezing are essential for proper enzyme assay.
- The vast majority of individuals with NKH have no detectable GCS activity.
- Individuals with a defect in the T-protein component (encoded by AMT) tend to have GCS activity up to 25% of normal values. Conversely, 50% of individuals with residual GCS activity in liver have AMT pathogenic variants [Toone et al 2003]. Individuals with a defect in the P-protein component (encoded by GLDC) do not tend to have residual GCS activity [Toone et al 2000] except for those who are mildly affected.
- Up to 5% of persons with deficient GCS activity do not have pathogenic variants in any NKH-related gene. These individuals could have pathogenic variants in genes encoding proteins involved in either GCS cofactors (lipoate and pyridoxal phosphate) or glycine transport (see Table 3, Differential Diagnosis).
Note: Although enzymatic confirmation of NKH using Epstein-Barr virus cultured lymphoblasts from peripheral blood samples was reported in six individuals with P-protein defects, others have obtained overlapping GCS activity in both controls and individuals with NKH, making this method unreliable [Applegarth et al 2000b].
Other testing (used in some locations):
- Glycine exchange reaction, which measures the combined activity of the P- and H-protein without the need for T-protein activity, can assist in identifying the specific subunit involved.
- 13C-glycine breath test shows decreased 13C-CO2 exhalation in individuals with NKH [Kure et al 2006b].
Clinical Characteristics
Clinical Description
Nonketotic hyperglycinemia (NKH) is the inborn error of glycine metabolism defined by deficient activity of the glycine cleavage enzyme system (GCS), which results in accumulation of large quantities of glycine in all body tissues including the brain.
NKH is categorized into severe NKH and attenuated NKH based on ultimate outcome [Swanson et al 2015]:
- Severe NKH. Children make no developmental progress and have intractable epilepsy.
- Attenuated NKH. Children make variable developmental progress and have treatable or no epilepsy. Attenuated NKH is further divided into:
- Attenuated poor. Children have a developmental quotient (DQ) of <20 and all have epilepsy.
- Attenuated intermediate. Children have a DQ of 20 to 50 and easily treatable epilepsy or no epilepsy.
- Attenuated good. Children have a DQ >50 and do not have epilepsy.
The majority of children with NKH present in the neonatal period or in early infancy, with only the mildest cases presenting in late infancy or childhood. Outcomes by age of onset are as follows:
- Neonatal onset. 85% have severe NKH and 15% have attenuated NKH.
- Infantile onset (i.e., >2 weeks). 50% have severe NKH and 50% have attenuated NKH [Hennermann et al 2012, Swanson et al 2015].
- Onset age >3 months. All had attenuated NKH.
Presentation
Neonatal (first hours to days of life) with progressive lethargy evolving into profound coma and marked hypotonia. In 80% of infants ventilatory drive slows, leading to prolonged apnea and often death if not supported by intubation and ventilation; in contrast, 20% of infants maintain spontaneous ventilation. The vast majority of infants regain spontaneous respiration within the first three weeks of life, and some show some spontaneous improvement in alertness in the first month of life, often with oral bottle drinking.
Myoclonic jerks and hiccups are often a sign of epilepsy. A history of prenatal hiccups is frequently present.
The initial EEG often shows a burst suppression pattern.
Infantile (age >2 weeks to 3 months). While these infants do not have lethargy and coma in the first days of life, they often have a history of hypotonia from early on. They present with developmental delay and infantile-onset seizures that can be mild or increasingly difficult to treat.
Late (age >3 months) is rare, is always associated with the attenuated form, and involves developmental delays and possible mild seizures.
Outcome
Severe NKH. Many infants never make any developmental gains, but some regain some skills such as spontaneous bottle feeding, looking, and smiling, particularly when treated with benzoate (see Management). At most, affected children learn to smile and roll from side to back or side to front (i.e., developmental age 6 weeks to 3 months). At about age three to four months they begin to lose skills such as bottle feeding. They do not learn to sit or grasp; they have limited interaction with their environment.
Before age six months children with severe NKH begin to develop progressive spasticity (hyperreflexia, distal hypertonicity, and positive Babinski signs) and cortical blindness (often with poor fixation and sometimes with roving eye movements). Most have swallowing dysfunction requiring tube feeding.
Increasingly difficult-to-treat seizures develop in the first year, usually requiring multiple anticonvulsants with incomplete seizure control. The EEG pattern can evolve into hypsarrhythmia and/or multifocal spikes.
Many develop scoliosis or hip dislocation often requiring surgical intervention (if indicated in the overall condition) in childhood or adolescence [Ramirez et al 2012].
Occasionally children with severe NKH have cleft palate or clubfeet [Hennermann et al 2012]. Some develop secondary microcephaly.
Attenuated NKH (with outcomes ranging from poor to intermediate to good). In general, children in this category make variable developmental progress. They can learn to walk, reach and grasp, use sign language, and interact with caregivers and attend special education classes. They have little spasticity.
They may develop a seizure disorder, which is often relatively easy to treat with either benzoate or dextromethorphan alone or with the addition of a single anticonvulsant [Van Hove et al 2005] (see Management).
Hyperactivity is common, often severe, and poorly responsive to interventions [Wiltshire et al 2000, Hennermann 2006].
Many have choreic movements, a good prognostic sign [Hennermann et al 2012].
They can have intermittent episodes of severe lethargy, often triggered by fever and infection (sometimes reported in the past as a "mild episodic form").
An adult experienced acute decompensation while on valproate (which is contraindicated) [Hall & Ringel 2004] (see Management).
- Poor outcome (DQ <20). Individuals in this category have manifestations intermediate between attenuated and severe NKH. Developmentally, they learn to grasp objects, usually are able to sit, and have limited interaction with some signs. Spasticity – which is less than that observed in severe NKH – is nonetheless noticeably present. Although epilepsy is usually controlled with one or two anticonvulsants, hypsarrhythmia that is not controlled with anticonvulsants portends a poor outcome.
- Intermediate outcome (DQ 20-50). Individuals in this category learn to walk and communicate with some speech but mostly sign language. They can grasp items purposefully and eat independently. They attend special education classes in school. Most have choreatic movements, and pronounced hyperactivity, often in bursts.
- Good outcome (DQ >50). Individuals in this category make substantial developmental progress and do not have epilepsy. Half of the individuals in this category present after age three months, with a few presenting after age one year. They sometimes can attend normal class in school. They have attention deficit and hyperactivity disorder (ADHD).They can have episodes of severe lethargy with infections [Brunel-Guitton et al 2011]. The recognition of the episodes of lethargy led to the description of the "mild episodic form," reported in four children with mild intellectual disability and episodes of chorea, agitated delirium, and vertical gaze palsy associated with febrile illness [Steiner et al 1996].Individuals homozygous for p.Ala802Val (which is associated with substantial residual GCS activity) who received early and aggressive treatment in the first two years of life had normal intelligence [Korman et al 2004] (see Management).
Other findings include the following [Authors, unpublished observations]:
- A number of affected individuals have had delayed gastric emptying and poor gastrointestinal motility, leading to very severe problems including dependency on total parenteral nutrition (TPN) in a few.
- A few individuals had sudden severe electrolyte disturbances including profound hypokalemia causing sudden cardiac arrest. This occurrence was rare (<1%) and did not recur.
- A few individuals have reported dysuria with difficulty emptying the bladder. It is unclear if this is a side effect of dextromethorphan or a manifestation of the disorder.
- In infants with severe NKH, a retrocerebellar cyst with subsequent development of hydrocephalus occurred in 3% of cases [Van Hove et al 2000], requiring ventriculoperitoneal shunt placement.
- Affected individuals can have recurrent and long episodes of unexplained severe crying.
Note: Some atypical manifestations historically reported as NKH (e.g., cardiomyopathy or with optic atrophy) are consistent with features of variant NKH (lipoate, iron-sulfur cluster defects) (see Differential Diagnosis), and not with classic NKH caused by deficient GCS activity due to biallelic pathogenic variants in AMT or GLDC.
Intrafamilial Variability
The phenotype of severe versus attenuated NKH is consistent within families, but the subcategory of attenuated NKH and degree of developmental progress can vary.
A retrospective study showed a consistent phenotype within seven families with two or more affected children [Hoover-Fong et al 2004]. The familial concordance for outcome has been observed in several additional families.
In sibs with significant variability in developmental outcome for attenuated NKH, aggressive treatment in the first two years of life with sodium benzoate and N-methyl D-aspartate (NMDA) receptor site antagonists was associated with improved developmental outcome [Korman et al 2004, Bjoraker et al 2016] (see Management).
Prognostic Predictors
Age at presentation. Individuals presenting later have attenuated disease; however, early presentation is not sufficiently predictive as 15% of individuals who present as neonates have attenuated disease.
Biochemically, plasma glycine alone does not predict developmental outcome. CSF glycine elevated >230 μmol/L predicts severe outcome; a CSF:plasma glycine ratio of <0.08 predicts attenuated outcome [Swanson et al 2015].
Radiologically, the presence of hydrocephalus predicts severe NKH; the presence of a very thin and shortened corpus callosum also predicts severe NKH [Van Hove et al 2000, Stence et al 2019].
The pattern of diffusion restriction on brain MRI is not predictive of phenotype [Stence et al 2019].
Clinically, the development of clear pyramidal tract signs before age six months predicts severe NKH, whereas the presence of choreatic movements predicts attenuated NKH [Hennermann et al 2012]. Attenuated NKH with a poor outcome can have signs intermediate between severe and attenuated outcome and early on can be difficult to distinguish clinically. Cleft palate and clubfeet when present predict severe outcome.
EEG. Persistent burst suppression pattern tends to be associated with severe outcome.
MRS. The glycine/creatine ratio is higher in severe than in attenuated NKH [Stence et al 2019].
Genotype. For genotypes that predict prognosis, see Genotype-Phenotype Correlations.
Genotype-Phenotype Correlations
There are no clinical differences between individuals with biallelic pathogenic variants in GLDC and those with pathogenic variants in AMT.
Glycine cleavage enzyme system (GCS) activity predicts severe versus attenuated outcome in NKH (see Clinical Description) [Swanson et al 2015] as follows:
- Biallelic pathogenic variants associated with lack of residual GCS activity, such as exon copy number variants, frameshift variants, nonsense variants, and consensus splice site variants (-1,2 or +1,2), have no residual activity except the following GLDC variants: c.2203-2A>G and c.2999delG (p.Cys1000LeufsTer31), a very late frameshift.
- Biallelic pathogenic variants with preserved residual GCS activity predict attenuated NKH, with the majority having attenuated good outcome.
- The presence of one variant with preserved residual GCS activity usually results in attenuated NKH, and on occasion results in severe NKH. In individuals with attenuated NKH, outcome ranges from attenuated poor to intermediate, with a few good.Note: The amount of residual activity detected in expression studies sufficient for attenuated outcome is as low as 1%.
- The residual function of a missense variant may be difficult to assess for the purpose of predicting genotype-phenotype correlation:
- Thus far, the expression of 47 missense GLDC variants has been reported [Swanson et al 2015, Bravo-Alonso et al 2017]. Commonly recurring variants are listed in Table 5.
- The expression of AMT variants has not yet been reported, but clinical studies implicate the very common pathogenic variant p.Arg320His as severe.
Nomenclature
Collectively, neurologic disorders caused by disturbance of glycine metabolism and transport are termed "glycine encephalopathy." See Differential Diagnosis for details about other inherited disorders causing glycine encephalopathy.
Note: The term "atypical NKH" is no longer used as it combined cases of attenuated NKH and variant NKH (see Table 3) and therefore was inconsistent and nonspecific. Many individuals described in the past as having atypical NKH (e.g., NKH with optic atrophy and progressive spasticity, NKH with cardiomyopathy, or NKH with pulmonary hypertension) are now known to have – or likely had – lipoate deficiency disorders.
Prevalence
The birth incidence of NKH has been estimated at 1:55,000 newborns in Finland (1:12,000 in an area of Northern Finland) and 1:63,000 in British Columbia, Canada [Applegarth et al 2000a]. The calculated carrier frequency is approximately 1:125 in the population of British Columbia, Canada (predominantly a population of northern European origin at the time of data collection for disease incidence). Using publicly available population genotypes, the birth estimate of NKH worldwide was estimated at 1:76,000 [Coughlin et al 2017]. An increased incidence is expected in populations with founder variants (see Table 5).
NKH may be underdiagnosed for several reasons:
- Attenuated NHK and severe NKH without apnea are clinically underappreciated (e.g., identification on exome sequencing in cases of autism [Yu et al 2013]).
- Analysis of CSF amino acids to detect elevated CSF glycine in infants with neonatal/infantile epilepsy, a primary trigger for suspicion of NKH, is not consistently obtained; furthermore, in NKH plasma glycine levels can be normal, and elevated levels are not specific for NKH.
- Multigene panels for neonatal/infantile epilepsy often do not include GLDC and AMT unless specifically requested.
Genetically Related (Allelic) Disorders
No phenotypes other than those discussed in this GeneReview are known to be associated with biallelic pathogenic variants in AMT or GLDC.
Although an association of heterozygous variants in GLDC and AMT with neural tube defects has been noted in two studies [Narisawa et al 2012, Shah et al 2016], neural tube defects have not been observed in parents of children with NKH.
Differential Diagnosis
Inherited disorders in the differential diagnosis of nonketotic hyperglycinemia (NKH) caused by deficient activity of the glycine cleavage enzyme system (GCS) are outlined in Table 3.
Table 3.
Inherited Disorders in the Differential Diagnosis of NKH
Clinical findings in the differential diagnosis of NKH. Of note, a single individual with glyceric aciduria with elevated glycine levels and deficient GCS enzyme activity was demonstrated to have co-occurrence of both conditions [Swanson et al 2017]. Subsequently co-occurrence of deficient GCS enzyme activity has not been identified in any other individuals with glyceric aciduria.
Hyperglycinemia. The differential diagnosis of nonketotic hyperglycinemia includes the following:
- Valproate treatment, which causes a secondary decrease in liver glycine cleavage enzyme system (GCS) activity and can reversibly increase the CSF glycine concentration to >60 µmol/L [Jaeken & Van Hove, unpublished observations]
- Hyperglycinemia of unknown cause identified on newborn screening (NBS). Several neonates have been reported to have isolated and persistently elevated serum glycine concentrations (>1,000 µmol/L) on NBS. CSF glycine concentration measured in one child was normal. Although the infants were asymptomatic, long-term follow up is not yet available. Molecular genetic testing for nonketotic hyperglycinemia did not identify causative variants in GLDC, AMT, or GCSH.
- Severe liver failure can cause hyperglycinemia (>2,000 μmol/L), which can precede other manifestations of liver failure. Of note, liver failure in neonates can result from acute herpes simplex virus infection.
- Loading with large amounts of glycine (e.g., use of immunoglobulins in a glycine buffer or bladder irrigation with a glycine-based solution) can cause substantial hyperglycinemia.
Transient glycine encephalopathy was the term used initially to describe the findings in neonates who presented with neonatal epileptic encephalopathy (i.e., seizures and/or burst suppression pattern on EEG) with biochemical findings of glycine encephalopathy that subsequently resolved over time. Some individuals had normal development; others had persistent neurologic impairment [Boneh et al 1996, Aliefendioğlu et al 2003]. All had normal GCS activity [Lang et al 2008] and no AMT, GLDC, or GCSH pathogenic variants.
Recent reports of causative intracerebral hemorrhage [Manley et al 2010] and hypoxic-ischemic injury [Aburahma et al 2011] indicate that transient glycine encephalopathy is a biochemical phenocopy of glycine encephalopathy. Unrecognized perinatal hypoxic-ischemic injury can present with progressive neonatal coma and seizures resulting from breakdown of the blood-brain barrier and seepage of serum into the CSF with elevation of CSF glycine concentration and elevated CSF:plasma glycine ratio, elevated CSF protein concentration, and often (small) elevations in several other amino acids in the CSF (particularly branched-chain amino acids). Note that on brain MRI the diffusion restriction in the posterior limb of the internal capsule observed in hypoxic-ischemic injury is not typically accompanied by diffusion restriction in the brain stem and cerebellum, findings that are characteristic of NKH [Stence et al 2019].
In the presence of hemorrhage, there can be elevated CSF glycine and elevated glycine on magnetic resonance spectroscopy [Manley et al 2010].
Hyperglycinuria. Severe asymptomatic hyperglycinuria can result from defects in the renal transport of glycine. Note: The previously reported association of the following conditions with symptoms likely reflects ascertainment bias [Coşkun et al 1993, Bröer et al 2008].
Inherited defects in renal transport of glycine:
- Familial iminoglycinuria. Homozygotes for a null allele in SLC36A2, the high-affinity transporter for proline, hydroxyproline, and glycine in the proximal renal tubule, have iminoglycinuria, whereas heterozygotes can have isolated glycinuria. Persons with a pathogenic variant in SLC36A2 and a benign variant in SLC6A18 may also have glycinuria [Bröer et al 2008].
- Benign hyperglycinuria. A common transient finding caused by immaturity of renal glycine reabsorption
MRI findings that have differential diagnostic implications particularly in the setting of elevated glycine levels include the following:
- Gyral malformations or true absence of the corpus callosum are not part of NKH, but have been noted in some cases of variant NKH [Ajit Bolar et al 2013] and pyridoxal phosphate disorders [Mills et al 2010].
- Cystic leukoencephalopathy would indicate possible variant NKH due to disorders in lipoate and/or iron-sulfur cluster metabolism. A mild increase of CSF glycine has also been reported in vanishing white matter disease [van der Knaap et al 1999].
Management
Evaluations Following Initial Diagnosis
To establish the extent of disease and needs in an individual diagnosed with nonketotic hyperglycinemia (NKH), the evaluations summarized in this section (if not performed as part of the evaluation that led to the diagnosis) are recommended.
- MRI of the brain in neonates. Brain MRI can reveal a retrocerebellar cystic area and development of hydrocephalus, which occurs in 3% of individuals with severe NKH [Van Hove et al 2000, Hoover-Fong et al 2004, Swanson et al 2015]. The reduction in length and particularly in thickness of the corpus callosum are predictive of outcome (shorter and thinner in severe NKH), and the growth rate of the thickness is worse in severe NKH than in attenuated NKH. Over time, brain atrophy develops in severe NKH.
- EEG. The presence of a burst suppression pattern, rather than multifocal seizures, is a poor prognostic indicator. Hypsarrhythmia is a background pattern associated with poor prognosis, unless early intervention resolves it.
- Developmental assessment throughout the first years of life, particularly in attenuated NKH
- Neurologic assessment in the first year to identify early development of spasticity in severely affected children and of chorea in those more mildly affected, and for the recognition and treatment of epilepsy
- Orthopedic evaluation in childhood in severe NKH for the presence of scoliosis and hip dislocation
- Pulmonary evaluation for poor breathing and respiratory hygiene predisposing to recurrent pulmonary infections in severe NKH. This is usually indicated in early childhood.
- Ophthalmologic assessment for developing cortical blindness. This is present in severe NKH and to some degree in attenuated NKH with a poor outcome. Visual evoked potentials can aid in the evaluation of cortical blindness.
- GI evaluation for gastrostomy tube feeding, obstipation, and occasionally dysmotility
- Consultation with a medical geneticist and/or genetic counselor
Treatment of Manifestations
No formal management guidelines have been developed for NKH.
Current treatment is focused on:
For severe NKH, no treatment is effective in changing the natural history of developmental delays, spasticity, and intractable epilepsy. Specifically, glycine-lowering therapy is not effective in improving the affected individual's development, even when initiated at birth [Korman et al 2006]. However, glycine-lowering therapy does decrease the frequency and severity of seizures and is used as part of the overall epilepsy management of disease [Hennermann et al 2012]. It also improves attentiveness and resolves neonatal apnea.
In contrast, for attenuated NKH current treatment consists of reduction of plasma concentration of glycine by treatment with sodium benzoate and blockade of NMDA receptors, which are overstimulated at the glycinergic site. To date two independent studies have shown that early, aggressive treatment of children with pathogenic variants associated with residual glycine cleavage enzyme system (GCS) activity who are likely to develop attenuated NKH resulted in improved neurodevelopmental outcome and reduced propensity for epilepsy [Korman et al 2004, Bjoraker et al 2016]. Further, in individuals with attenuated NKH, sodium benzoate improves alertness, reduces or eliminates episodic lethargy, and may also improve behavior.
Reduction of Plasma Concentration of Glycine
Sodium benzoate can reduce the plasma glycine concentration into the normal range (Table 1). The therapeutic goal is to lower the plasma glycine concentration into the low normal range, defined as 120 to 300 µmol/L for samples obtained one to two hours after a benzoate dose (timing is important).
The dose required depends on the glycine pool available. *
- Individuals with attenuated NKH require a lower dose (200-550 mg/kg/day). For older children and adults, consider dosing based on body surface area (e.g., for attenuated NKH start at 5.5 g/m2 BSA).
- Individuals with severe NKH require a higher dose (550-750 mg/kg/day) [Van Hove et al 2005]; for adults, maximum 16.5 g/m2/day.
* Note: Because the glycine pool is reduced when individuals are on a ketogenic diet, sodium benzoate dose must be reduced upon initiation of this diet to avoid toxicity.
Sodium benzoate should be divided into no less than three doses per day; doses are more frequent in infancy (for example, neonates typically receive six daily doses).
Benzoate treatment begins with the lower dose range for the predicted disease severity; plasma glycine concentration is measured regularly. If the plasma glycine concentration is not within target range, the dose is increased by 50 mg/kg/day, and plasma glycine concentration is measured again as soon as 24 to 48 hours later. When glycine is within the target range, plasma glycine levels are measured regularly: every two weeks for infants, every month for young children, and every three months for older children.
Because the liver and kidney (but not the brain) are the sites of action of sodium benzoate, it is unclear to what extent administration of sodium benzoate reduces brain or CSF glycine. It is known that treatment with sodium benzoate does not normalize CSF glycine concentration. Follow up with serial measurements of CSF glycine concentration is not required.
Side effects of sodium benzoate include the following:
- High-dose sodium benzoate (500-750 mg/kg/day) is frequently associated with gastritis, which may require oral administration of antacids, H2 antagonists, or proton pump inhibitors.
- High-dose sodium benzoate in young infants can be associated with excessive loss of carnitine; those with low carnitine levels should receive supplementation to maintain normal plasma concentrations.
- Dosing of sodium benzoate in excess of the individual requirement is dangerous: benzoate toxicity has high morbidity and mortality [Van Hove et al 2005]. Hypocalcemia and low plasma glycine concentration (<150 µmol/L) can be early signs of sodium benzoate overdose. Measurement of plasma benzoate concentration can be helpful in evaluating potential toxicity (toxicity >2.5 mmol/L). Benzoate toxicity is treated by withholding benzoate, giving glycine, and/or hemodialysis.
- As benzoate is unpalatable, a saliva-resistant granulated benzoate is available in several countries for individuals not on tube feeding. When transferring individuals from regular benzoate to granulated benzoate, providers should consider that the benzoate content in the granulated form is approximately 75%.
Glycine-restricted diet. In NKH, the contribution of dietary glycine is small compared to the excess in endogenous glycine synthesis versus endogenous catabolism of glycine. Infant formula is typically low in glycine; advancing the diet to intake of solid food introduces a small amount of extra dietary glycine. Restriction of dietary glycine can aid in controlling plasma glycine levels for some individuals with severe NKH. For many individuals a mild increase in the dose of sodium benzoate compensates for increased dietary intake of glycine.
An inappropriately severe glycine-restricted diet has been associated with protein malnutrition [Rogers et al 2014]; thus, the limited benefits of glycine restriction often do not outweigh the associated complexity and risk.
NMDA Receptor Site Antagonists
Glycine is an allosteric activator of the NMDA receptor channel complex; thus, excess glycine can result in overstimulation, which has been putatively linked to seizures and developmental delays. Clinically used partial inhibitors of the NMDA receptor include dextromethorphan, ketamine, or felbamate.
Dextromethorphan doses commonly range from 3 to 15 mg/kg/day, but individual variability is substantial. The authors have typically started at 10 mg/kg/day for neonates, 5 mg/kg/day for children, and 3 mg/kg/day for adolescents and adults, administered in three or four doses per day, or in twice-daily dosing if using a slow-release version (e.g., Delsym® Extended Release).
Pharmacogenomic differences exist in the metabolism of dextromethorphan, particularly based on CYP2D6 polymorphism. Some concomitant medications may slow the metabolism of dextromethorphan (e.g., cimetidine) and should be reviewed or not used as they may cause toxicity [Arnold et al 1997]. The effect of this in the treatment of NKH has not been reviewed. Blood concentration can technically be monitored; however, since the therapeutic level is not defined (should be >0 and <100 nmol/L), there is currently no clinically utility, and it is performed in a research context only [Hamosh et al 1998]. Overdose of dextromethorphan can cause increases in sleepiness and movement.
Treatment effect:
- Attenuated NKH. Dextromethorphan used in combination with sodium benzoate has improved neurocognitive outcome and decreased seizure propensity. Improved attention, school performance, and behavior, as well as decreased chorea, have been observed in several individuals with attenuated NKH [Authors, personal observation]. In itself, high-dose dextromethorphan may have some anticonvulsant activity.
- Severe NKH. The effect of dextromethorphan in severe NKH is dubious. Furthermore, the use of dextromethorphan in severe NKH is associated with a higher rate of pneumonia.
Oral ketamine has been used in NKH as an NMDA receptor antagonist. Improvement in outcome has been documented in attenuated NKH.
Glycinergic inhibitory receptors. Although strychnine improves tone and respiration, its use has been abandoned because of serious side effects that result from its long-term use.
Symptomatic Treatment
Seizure control. A systematic review of epileptic phenotypes, EEG patterns, and response to anti-seizure medications has rarely been done [Hennermann et al 2012].
Control of seizure disorders associated with a severely disturbed background such as burst suppression pattern or hypsarrhythmia is essential to allow developmental progress. Control tends to be challenging in severe NKH but is usually possible in attenuated NKH and essential for good outcome:
- Severe NKH. Epilepsy propensity worsens in the first year of life, and from the second year of life, individuals have intractable epilepsy (i.e., daily seizures despite treatment with ≥2 anticonvulsants).
- Attenuated NKH. First-line treatment is reduction of glycine levels with benzoate and dextromethorphan. This combination results in improvement of EEG background and reduced seizures; many individuals with attenuated NKH do not experience seizures on this treatment.
First-line treatment for newborns and infants with myoclonic seizures is benzodiazepines, with clobazam currently the preferred first-line drug, whereas older literature mentioned clonazepam and diazepam. Variable results are reported with use of standard anti-seizure medication (ASM) in neonates. Phenytoin has limited efficacy for seizure control. The effect of phenobarbital is variable in neonates, but because the nature of the epilepsy changes in late infancy, phenobarbital is often useful in treating seizures in older affected children.
Other drugs used with variable effect include levetiracetam and topiramate. Various ASMs have been used with variable success. Felbamate has been successful in some children with difficult-to-treat seizures. This treatment must be closely monitored for signs of liver or hematopoietic toxicity.
Ketogenic diet has been used in some individuals with variable success. Ketogenic diet always lowers the amount of glycine substantially and the dose of sodium benzoate should be reduced accordingly to avoid benzoate toxicity [Cusmai et al 2012]. Ketogenic diet has resulted in improved seizure control, but did not change hypsarrhythmic background.
For some older individuals with severe NKH and difficult-to-control seizures, a vagal nerve stimulator has been used with varying (sometimes very high) levels of success [Tsao 2010].
Treatment of infantile spasms and hypsarrhythmia in the context of severe NKH is difficult. Steroids rarely have an effect; vigabatrin has resulted in loss of skills and adverse outcome in individuals with attenuated NKH [Tekgul et al 2006; Authors, personal observation]. Glycine reduction with sodium benzoate, dextromethorphan, and other anticonvulsants has been the best approach for infantile spasms and hypsarrhythmia in attenuated NKH, whereas for severe NKH, treatment is difficult and conventional ASMs and ketogenic diet appear to have the best result – albeit often with only limited success.
Other. Gastrostomy tube placement should be considered early in the management of individuals with swallowing dysfunction associated with severe disease. Gastroesophageal reflux is common. Some individuals have benefited from a Nissen procedure. Chronic obstipation, a frequent problem in severe NKH, can be treated with laxatives.
Most affected individuals need physical therapy.
Scoliosis and hip dislocation, common in older children with severe NKH, are managed with standard techniques. The utility of these procedures has to be weighted in the quality of life of the individual.
Individuals with severe NKH have progressive difficulty maintaining good airway management. Pulmonary review and assistance can greatly facilitate quality of life.
Withdrawal of Intensive Care Support
Up to 80% of neonates with symptomatic NKH develop life-threatening bradypnea or apnea and require ventilator assistance during the first week of life. In the second to the third week of life spontaneous breathing typically resumes (even in the absence of treatment to reduce glycine levels), allowing discontinuation of ventilator assistance. Following resumption of spontaneous breathing, apnea is unlikely to recur. Following resolution of the apneic phase, some untreated infants with neonatal-onset NKH may die in the next two years, but many – if not most – live for several years.
Because of the generally poor prognosis of neonatal-onset NKH, some families elect to withdraw intensive care support during the neonatal apneic phase, allowing the infant to succumb prior to recovery of spontaneous respiration. For a discussion of the ethics involved in deciding to withdraw support for neonates with apnea see Boneh et al [2008].
Surveillance
Developmental assessment should be performed throughout the first years of life.
Neurologic assessments in the first year can identify early development of spasticity in severely affected individuals and early development of chorea in more mildly affected individuals.
Severely affected individuals should be monitored for scoliosis and hip dysplasia.
Pulmonary function should be assessed, particularly in children who develop recurrent respiratory infections.
Agents/Circumstances to Avoid
Valproate is contraindicated in NKH as an anti-seizure medication. It raises blood and CSF glycine concentrations and may increase seizure frequency. It has resulted in severe lethargy, coma, severe seizures, and chorea particularly in mildly affected individuals [Hall & Ringel 2004; Authors, personal observation].
Vigabatrin has resulted in rapid loss of function when used to treat West syndrome in NKH caused by deficient activity of the glycine cleavage enzyme system [Tekgul et al 2006].
Evaluation of Relatives at Risk
Biochemical testing to promote early diagnosis and treatment of at-risk newborn sibs is indicated. In particular, sibs at risk for attenuated NKH can benefit from early and aggressive treatment [Korman et al 2004, Bjoraker et al 2016].
If the pathogenic variants in the family are known, biochemical testing can be followed with molecular genetic testing.
See Genetic Counseling for issues related to testing of at-risk relatives for genetic counseling purposes.
Pregnancy Management
Pregnancy has been reported in one woman with attenuated NKH. No obvious teratogenic effect was observed and intelligence was normal [Ellaway et al 2001].
Therapies Under Investigation
Search ClinicalTrials.gov in the US and EU Clinical Trials Register in Europe for access to information on clinical studies for a wide range of diseases and conditions. Note: There may not be clinical trials for this disorder.
Genetic Counseling
Genetic counseling is the process of providing individuals and families with information on the nature, mode(s) of inheritance, and implications of genetic disorders to help them make informed medical and personal decisions. The following section deals with genetic risk assessment and the use of family history and genetic testing to clarify genetic status for family members; it is not meant to address all personal, cultural, or ethical issues that may arise or to substitute for consultation with a genetics professional. —ED.
Mode of Inheritance
Nonketotic hyperglycinemia (NKH) is inherited in an autosomal recessive manner.
Risk to Family Members
Parents of a proband
- The parents of an affected individual are typically heterozygotes (i.e., carriers of one NKH-related pathogenic variant).
- Heterozygotes are asymptomatic and are not at risk of developing the disorder.
- De novo pathogenic variants occur in approximately 1% of individuals with NKH; thus, carrier status in parents should be confirmed by molecular genetic testing rather than be assumed or inferred [Swanson et al 2015, Coughlin et al 2017].
Sibs of a proband
- If both parents are heterozygous for one pathogenic variant, each sib of an affected individual has at conception a 25% chance of being affected, a 50% chance of being an asymptomatic carrier, and a 25% chance of being unaffected and not a carrier.
- Heterozygotes (carriers) are asymptomatic and are not at risk of developing the disorder.
Offspring of a proband. Pregnancy has been reported in one woman with attenuated NKH (see Pregnancy Management); the offspring of an affected individual are obligate heterozygotes (carriers) for a pathogenic variant in an NKH-related gene.
Other family members. Each sib of the proband's parents is at a 50% risk of being a carrier of an NKH-related pathogenic variant.
Carrier Detection
Molecular genetic testing. Carrier testing for at-risk relatives requires prior identification of the NKH-related pathogenic variants in the family.
Note: Carrier testing using biochemical methods is not reliable.
Related Genetic Counseling Issues
See Management, Evaluation of Relatives at Risk for information on evaluating at-risk relatives for the purpose of early diagnosis and treatment.
Family planning
- The optimal time for determination of genetic risk, clarification of carrier status, and discussion of the availability of prenatal/preimplantation genetic testing is before pregnancy.
- It is appropriate to offer genetic counseling (including discussion of potential risks to offspring and reproductive options) to young adults who are affected, are carriers, or are at risk of being carriers.
DNA banking. Because it is likely that testing methodology and our understanding of genes, pathogenic mechanisms, and diseases will improve in the future, consideration should be given to banking DNA from probands in whom a molecular diagnosis has not been confirmed (i.e., the causative pathogenic mechanism is unknown). For more information, see Huang et al [2022].
Prenatal Testing and Preimplantation Genetic Testing
Molecular genetic testing. Once the pathogenic variants in an NKH-related gene have been identified in an affected family member, prenatal testing for a pregnancy at increased risk and preimplantation genetic testing are possible.
If only a single pathogenic variant in a causative gene is known, and if material from the proband and both parents is available, intragenic SNPs have been used to provide intragenic linkage analysis.
Enzymatic testing. When prenatal testing by molecular means is not possible (e.g., molecular genetic alteration has not been identified), prenatal testing by assay of the GCS enzyme activity in uncultured CVS material is possible. Prenatal diagnosis by GCS enzyme assay has at least a 1% false negative rate [Applegarth et al 2000b]. False negative results appear particularly to involve T-protein deficiency in individuals with attenuated NKH in whom the range of enzyme activity in affected individuals and heterozygotes (carriers) overlaps.
Resources
GeneReviews staff has selected the following disease-specific and/or umbrella support organizations and/or registries for the benefit of individuals with this disorder and their families. GeneReviews is not responsible for the information provided by other organizations. For information on selection criteria, click here.
- Joseph’s GoalUnited KingdomEmail: [email protected]
- MedlinePlus
- NKH CrusadersPhone: 781-249-1835Email: [email protected]
- NKH Deutsches Familien NetzwerkGermany
- NKH International Family Network2236 Birchbark TrailClearwater FL 33763Phone: 727-799-4977Fax: 727-441-4942Email: [email protected]
Molecular Genetics
Information in the Molecular Genetics and OMIM tables may differ from that elsewhere in the GeneReview: tables may contain more recent information. —ED.
Table A.
Nonketotic Hyperglycinemia: Genes and Databases
Table B.
OMIM Entries for Nonketotic Hyperglycinemia (View All in OMIM)
Molecular Pathogenesis
Nonketotic hyperglycinemia (NKH) is an inborn error of glycine metabolism caused by deficient activity of the glycine cleavage enzyme system (GCS) (Figure 2).
GLDC encodes glycine decarboxylase or P-protein, a pyridoxal 5'-phosphate-containing homodimer. It completes the first of four steps in glycine degradation (see Figure 2), by P-protein-catalyzed decarboxylation of glycine with CO2 as product.
AMT encodes aminomethyltransferase or T-protein, which requires tetrahydrofolate (THF) as cofactor. It transfers the methyl group from glycine to tetrahydrofolate and is the second step of the GCS.
GCSH encodes H-protein, a lipoamide-containing non-enzyme protein that interacts as a cosubstrate with all three enzyme proteins by providing the central arm for substrate binding on which the glycine cleavage cycle depends.
Mechanism of disease causation. NKH occurs through a loss-of-function mechanism. Residual enzyme activity correlates with the outcome of the disease (see Genotype-Phenotype Correlations).
Gene-specific laboratory technical considerations. See Table 4.
Table 4.
Laboratory Technical Considerations for Genes Causing Nonketotic Hyperglycinemia
More than 420 unique pathogenic variants were deposited in the Leiden Open Variant Database (LOVD) [Coughlin et al 2017]. Select recurring pathogenic variants and the founder variants in certain populations are listed in Table 5 [Swanson et al 2015, Coughlin et al 2017].
Table 5.
Notable Pathogenic Variants in Genes Causing Nonketotic Hyperglycinemia
Chapter Notes
Author History
Derek A Applegarth, PhD, FCCMG; University of British Columbia (2002-2005)
Curtis Coughlin II, MS, MBe (2013-present)
Ada Hamosh, MD, MPH; Johns Hopkins University School of Medicine (2002-2013)
Julia B Hennermann, MD, PhD (2019-present)
S Lane Rutledge, MD; University of Alabama at Birmingham (2018-2019; see Acknowledgments)
Gunter Scharer, MD, PhD (2009-2013)
Michael A Swanson, PhD (2019-present)
Jennifer Toone, BSc, RT; Children's and Women's Health Centre of British Columbia (2002-2005)
Johan Van Hove, MD, PhD (2009-present)
Acknowledgments
The authors would like to dedicate this GeneReview to Dr S Lane Rutledge, MD. Dr Rutledge cared for many patients with NKH over many years. She was compassionate, dedicated, and with a keen sense of what was important to families and children. She contributed to this version with many practical insightful comments. She was taken from us suddenly in 2019. She is missed by her patients and colleagues.
Revision History
- 23 May 2019 (bp) Comprehensive update posted live
- 11 July 2013 (me) Comprehensive update posted live
- 24 November 2009 (me) Comprehensive update posted live
- 26 July 2005 (ah) Revision: molecular genetic testing clinically available
- 14 December 2004 (me) Comprehensive update posted live
- 16 May 2003 (cd) Revision: enzymatic prenatal testing no longer available
- 14 November 2002 (me) Review posted live
- 7 March 2002 (da) Original submission
References
Literature Cited
- Aburahma S, Khassawneh M, Griebel M, Sharp G, Gibson J. Pitfalls in measuring cerebrospinal fluid glycine levels in infants with encephalopathy. J Child Neurol. 2011;26:703–6. [PubMed: 21335543]
- Ajit Bolar N, Vanlander AV, Wilbrecht C, Van der Aa N, Smet J, De Paepe B, Vandeweyer G, Kooy F, Eyskens F, De Latter E, Delanghe G, Govaert P, Leroy JG, Loeys B, Lill R, Van Laer L, Van Coster R. Mutation of the iron-sulfur cluster assembly gene IBA57 causes severe myopathy and encephalopathy. Hum Mol Genet. 2013;22:2590–602. [PubMed: 23462291]
- Aliefendioğlu D, Tana Aslan A, Coşkun T, Dursun A, Cakmak FN, Kesimer M. Transient nonketotic hyperglycinemia: two case reports and literature review. Pediatr Neurol. 2003;28:151–5. [PubMed: 12699870]
- Applegarth DA, Edelstein AD, Wong LT, Morrison BJ. Observed range of assay values for plasma and cerebrospinal fluid amino acid levels in infants and children aged 3 months to 10 years. Clin Biochem. 1979;12:173–8. [PubMed: 519849]
- Applegarth DA, Toone JR. Nonketotic hyperglycinemia (glycine encephalopathy): laboratory diagnosis. Mol Genet Metab. 2001;74:139–46. [PubMed: 11592811]
- Applegarth DA, Toone JR, Lowry RB. Incidence of inborn errors of metabolism in British Columbia 1969-1996. Pediatrics. 2000a;105:e10. [PubMed: 10617747]
- Applegarth DA, Toone JR, Rolland MO, Black SH, Yim DK, Bemis G. Non-concordance of CVS and liver glycine cleavage enzyme in three families with non-ketotic hyperglycinaemia (NKH) leading to false negative prenatal diagnoses. Prenat Diagn. 2000b;20:367–70. [PubMed: 10820402]
- Arnold GL, Griebel ML, Valentine JL, Koroma DM, Kearns GL. Dextromethorphan in nonketotic hyperglycinaemia: metabolic variation confounds the dose-response relationship. J Inherit Metab Dis. 1997;20:28–38. [PubMed: 9061564]
- Baker PR, Friederich MW, Swanson MA, Shaikh T, Bhattacharya K, Scharer GH, Aicher J, Creadon-Swindell G, Geiger E, Maclean KN, Lee WT, Deshpande C, Freckmann ML, Shih LY, Wasserstein M, Rasmussen MB, Lund AM, Procopis P, Cameron JM, Robinson BH, Brown GK, Brown RM, Compton AG, Dieckmann CL, Collard R, Coughlin CR, Spector E, Wempe MF, Van Hove JLK. Variant non ketotic hyperglycinemia is caused by mutations in LIAS, BOLA3 and the novel gene GLRX5. Brain. 2014;137:366–79. [PMC free article: PMC3914472] [PubMed: 24334290]
- Bjoraker KJ, Swanson MA, Coughlin CR 2nd, Christodoulou J, Tan ES, Fergeson M, Dyack S, Ahmad A, Friederich MW, Spector EB, Creadon-Swindell G, Hodge MA, Gaughan S, Burns C, Van Hove JL. Neurodevelopmental outcome and treatment efficacy of benzoate and dextromethorphan in siblings with attenuated nonketotic hyperglycinemia. J Pediatr. 2016;170:234–9. [PubMed: 26749113]
- Boneh A, Degani Y, Harari M. Prognostic clues and outcome of early treatment of nonketotic hyperglycinemia. Pediatr Neurol. 1996;15:137–41. [PubMed: 8888048]
- Boneh A, Korman SH, Sato K, Kanno J, Matsubara Y, Lerer I, Ben-Neriah Z, Kure S. A single nucleotide substitution that abolishes the initiator methionine codon of the GLDC gene is prevalent among patients with glycine encephalopathy in Jerusalem. J Hum Genet. 2005;50:230–4. [PubMed: 15864413]
- Boneh A, Allan S, Mendelson D, Spriggs M, Gillam L H, Korman S H. Clincial, ethical and legal considerations in the treatment of newborns with non-ketotic hyperglycinaemia. Mol Genet Metab. 2008;94:143–7. [PubMed: 18395481]
- Bravo-Alonso I, Navarrete R, Arribas L, Perona A, Abia D, Couce ML, Garcia-Cazorla A, Morais A, Domingo R. garcia A, Ramos M A, Van Hove J LK, Ugarte M, Pérez B, Pérez-Cerdá C, Rodríguez-Pombo P. Glycine encephalopathy: functional assessment of missense variants in GLDC gene to understand the phenotypes of the disease. Hum Mutat. 2017;38:678–91. [PubMed: 28244183]
- Bröer S, Balley CG, Kowalczuk S, Ng C, Vanslambrouck JM, Rodgers H, Auray-Blais C, Cavanaugh JA, Bröer A, Rasko JE. Iminoglycinuria and hyperglycinuria are discrete human phenotypes resulting from complex mutations in proline and glycine transporters. J Clin Invest. 2008;118:3881–92. [PMC free article: PMC2579706] [PubMed: 19033659]
- Brunel-Guitton C, Casey B, Coulter-Mackie M, Vallance H, Hewes D, Stockler-Ipsiroglu S, Mercimek-Mahmutoglu S. Late-onset nonketotic hyperglycinemia caused by a novel homozygous missense mutation in the GLDC gene. Mol Genet Metab. 2011;103:193–6. [PubMed: 21411353]
- Clayton PT, Surtees RAH, DeVile C, Hyland K, Heales SJR. Neonatal epileptic encephalopathy. Lancet. 2003;361:1614. [PubMed: 12747882]
- Coşkun T, Ozalp I, Tokatly A. Iminoglycinuria: a benign type of inherited aminoaciduria. Turk J Pediatr. 1993;35:121–5. [PubMed: 7504361]
- Coughlin CR 2nd, Swanson MA, Kronquist K, Acquaviva C, Hutchin T, Rodríguez-Pombo P, Väisänen ML, Spector E, Creadon-Swindell G, Brás-Goldberg AM, Rahikkala E, Moilanen JS, Mahieu V, Matthijs G, Bravo-Alonso I, Pérez-Cerdá C, Ugarte M, Vianey-Saban C, Scharer GH, Van Hove JL. The genetic basis of classical nonketotic hyperglycinemia due to mutations in GLDC and AMT. Genet Med. 2017;2017;19:104–11. [PubMed: 27362913]
- Cusmai R, Martinelli D, Moavero R, Dionisi Vici C, Vigevano F, Castana C, Elia M, Bernabei S, Bevivino E. Ketogenic diet in early myoclonic encephalopathy due to non ketotic hyperglycinemia. Eur J Paediatr Neurol. 2012;16:509–13. [PubMed: 22261077]
- Ellaway CJ, Mundy H, Lee PJ. Successful pregnancy outcome in atypical hyperglycinaemia. J Inherit Metab Dis. 2001;24:599–600. [PubMed: 11757588]
- Gabis L, Parton P, Roche P, Lenn N, Tudorica A, Huang W. In vivo 1H magnetic resonance spectroscopic measurement of brain glycine levels in nonketotic hyperglycinemia. J Neuroimaging. 2001;11:209–11. [PubMed: 11296595]
- Hall DA, Ringel SP. Adult nonketotic hyperglycinemia (NKH) crisis presenting as severe chorea and encephalopathy. Mov Disord. 2004;19:485–6. [PubMed: 15077252]
- Hamosh A, Maher J F, Bellus G A, Rasmussen S A, Johnston M V. Long-term use of high-dose benzoate and dextromethorphan for the treatment of nonketotic hyperglycinemia. J Pediatr. 1998;132:709–13. [PubMed: 9580775]
- Hayasaka K, Tada K. Effects of the metabolites of the branched-chain amino acids and cysteamine on the glycine cleavage system. Biochem Int. 1983;6:225–30. [PubMed: 6679320]
- Heindel W, Kugel H, Roth B. Noninvasive detection of increased glycine content by proton MR spectroscopy in the brains of two infants with nonketotic hyperglycinemia. AJNR Am J Neuroradiol. 1993;14:629–35. [PMC free article: PMC8333375] [PubMed: 8517351]
- Hennermann JB. Clinical variability in glycine encephalopathy. Future Neurol. 2006;1:621–30.
- Hennermann JB, Berger JM, Grieben U, Scharer G, Van Hove JL. Prediction of long-term outcome in glycine encephalopathy: a clinical survey. J Inherit Metab Dis. 2012;35:253–61. [PubMed: 22002442]
- Hoffmann G F, Schmitt B, Windfuhr M, Wagner N, Strehl H, Bagci S, Franz AR, Mills PB, Clayton PT, Baumgartner MR, Steinmann B, Bast T, Wolf NI, Zschocke J. Pyridoxal 5'-phosphate may be curative in early-onset epileptic encephalopathy. J Inherit Metab Dis. 2007;30:96–9. [PubMed: 17216302]
- Hoover-Fong JE, Shah S, Van Hove JL, Applegarth D, Toone J, Hamosh A. Natural history of nonketotic hyperglycinemia in 65 patients. Neurology. 2004;63:1847–53. [PubMed: 15557500]
- Huang SJ, Amendola LM, Sternen DL. Variation among DNA banking consent forms: points for clinicians to bank on. J Community Genet. 2022;13:389–97. [PMC free article: PMC9314484] [PubMed: 35834113]
- Jaeken J, de Koning T, van Hove J. Disorders of GABA, glycine, serine and proline. In: Blau N, Duran M, Blaskovics ME, Gibson KM, eds. Physician's Guide to the Laboratory Diagnosis of Metabolic Diseases. 2 ed. Berlin: Springer; 2002:123-40.
- Korman SH, Boneh A, Ichinohe A, Kojima K, Sato K, Ergaz Z, Gomori JM, Gutman A, Kure S. Persistent NKH with transient or absent symptoms and a homozygous GLDC mutation. Ann Neurol. 2004;56:139–43. [PubMed: 15236413]
- Korman SH, Wexler ID, Gutman A, Rolland MO, Kanno J, Kure S. Treatment from birth of nonketotic hyperglycinemia due to a novel GLDC mutation. Ann Neurol. 2006;59:411–5. [PubMed: 16404748]
- Kure S, Ichinohe A, Kojima K, Sato K, Kizaki Z, Inoue F, Yamanaka C, Matsubara Y. Mild variant of nonketotic hyperglycinemia with typical neonatal presentations: mutational and in vitro expression analyses in two patients. J Pediatr. 2004;144:827–9. [PubMed: 15192636]
- Kure S, Kato K, Dinopoulos A, Gail C, DeGrauw TJ, Christodoulou J, Bzduch V, Kalmanchey R, Fekete G, Trojovsky A, Plecko B, Breningstall G, Tohyama J, Aoki Y, Matsubara Y. Comprehensive mutation analysis of GLDC, AMT, and GCSH in nonketotic hyperglycinemia. Hum Mutat. 2006a;27:343–52. [PubMed: 16450403]
- Kure S, Korman SH, Kanno J, Narisawa A, Kubota M, Takayanagi T, Takayanagi M, Saito T, Matsui A, Kamada F, Aoki Y, Ohura T, Matsubara Y. Rapid diagnosis of glycine encephalopathy by 13C-glycine breath test. Ann Neurol. 2006b;59:862–7. [PubMed: 16634033]
- Kure S, Takayanagi M, Kurihara Y, Leisti J, Zalai D, Chuck G, Tada K, Matsubara Y, Narisawa K. Nonketotic hyperglycinemia: mutation spectra of the GLDC and AMT genes in Finnish and non-Finnish populations. Am J Hum Genet. 1999;65:A2406.
- Kure S, Takayanagi M, Narisawa K, Tada K, Leisti J. Identification of a common mutation in Finnish patients with nonketotic hyperglycinemia. J Clin Invest. 1992;90:160–4. [PMC free article: PMC443076] [PubMed: 1634607]
- Lang TF, Parr JR, Matthews EE, Gray RG, Bonham JR, Kay JD. Practical pitfalls in the diagnosis of transient non-ketotic hyperglycinemia. Dev Med Child Neurol. 2008;50:157–9. [PubMed: 18201306]
- Mayr JA, Feichtinger RG, Tort F, Ribes A, Sperl W. Lipoic acid synthesis defects. J Inherit Metab Dis. 2014;37:553–63. [PubMed: 24777537]
- Manley BJ, Sokol J, Cheong JLY. Intracerebral blood and MRS in neonatal nonketotic hyperglycinemia. Pediatr Neurol. 2010;42:219–22. [PubMed: 20159434]
- Mills PB, Surtees RA, Champion MP, Beesley CE, Dalton N, Scambler PJ, Heales SJ, Briddon A, Scheimberg I, Hoffmann GF, Zschocke J, Clayton PT. Neonatal epileptic encephalopathy caused by mutations in the PNPO gene encoding pyridox(am)ine 5'-phosphate oxidase. Hum Mol Genet. 2005;14:1077–86. [PubMed: 15772097]
- Mills PB, Footitt EJ, Mills KA, Tuschl K, Aylett S, Varadkar S, Hemingway C, Marlow N, Rennie J, Baxter P, Dular O, Nabout R, Craigen WJ, Schmitt B, Feillet F, Christensen E, De Lonlay P, Pike MG, Hughes MI, Struys EA, Jakobs C, Zuberi SM, Clayton PT. Genotypic and phenotypic spectrum of pyridoxine-dependent epilepsy (ALDH7A1 deficiency). Brain. 2010;133:2148–59. [PMC free article: PMC2892945] [PubMed: 20554659]
- Narisawa A, Komatsuzaki S, Kikuchi A, Niihori T, Aoki Y, Fiujiwara K, Tanemura M, Hata A, Suzuki Y, Relton CL, Grinham J, Leung KY, Pattidge D, Robinson A, Stone V, Gustavsson P, Stanier P, Copp AJ, Greene ND, Tominaga T, Matsubara Y, Kure S. Mutations in genes encoding the glycine cleavage system predispose to neural tube defects in mice and humans. Hum Mol Genet. 2012;21:1496–503. [PMC free article: PMC3298276] [PubMed: 22171071]
- Ramirez N, Flynn JM, Casalduc F, Rodriguez S, Cornier AS, Carlo S. Musculoskeletal manifestations of neonatal nonketotic hyperglycinemia. J Child Orthop. 2012;6:199–203. [PMC free article: PMC3400000] [PubMed: 23814620]
- Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, Grody WW, Hegde M, Lyon E, Spector E, Voelkerding K, Rehm HL, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17:405–24. [PMC free article: PMC4544753] [PubMed: 25741868]
- Rogers AS, Shaughnessy KK, Davis LS. Dermatitis and dangerous diets: a case of kwashiorkor. JAMA Dermatol. 2014;150:910–1. [PubMed: 24807070]
- Scalais E, Osterheld E, Weitzel C, De Meirleir L, Mataigne F, Martens G, Shaikh TH, Coughlin CR 2nd, Yu HC, Swanson M, Friederich MW, Scharer G, Helbling D, Wendt-Andrae J, Van Hove JLK. X-linked Cobalamin Disorder (HCFC1) Mimicking nonketotic hyperglycinemia with increased both cerebrospinal fluid glycine and methylmalonic acid. Pediatr Neurol. 2017;71:65–9. [PubMed: 28363510]
- Shah RH, Northrup H, Hixson JE, Morrison AC, Au KS. Genetic association of the glycine cleavage system genes and myelomeningocele. Birth Defects Res A Clin Mol Teratol. 2016;106:847–53. [PMC free article: PMC5074854] [PubMed: 27620832]
- Steiner RD, Sweetser DA, Rohrbaugh JR, Dowton SB, Toone JR, Applegarth DA. Nonketotic hyperglycinemia: atypical clinical and biochemical manifestations. J Pediatr. 1996;128:243–6. [PubMed: 8636821]
- Stence NV, Fenton LZ, Levek C, Tong S, Coughlin CR II, Hennermann J, Van Hove JLK. Brain imaging in classic nonketotic hyperglycinemia: quantitative analysis and relation to phenotype. J Inherit Metab Dis. 2019;42:438–50. [PubMed: 30737808]
- Swanson MA, Coughlin CR Jr, Scharer GH, Szerlong HJ, Bjoraker KJ, Spector EB, Creadon-Swindell G, Mahieu V, Matthijs G, Hennermann JB, Applegarth DA, Toone JR, Tong S, Williams K, Van Hove JL. Biochemical and molecular predictors for prognosis in nonketotic hyperglycinemia. Ann Neurol. 2015;78:606–18. [PMC free article: PMC4767401] [PubMed: 26179960]
- Swanson MA, Garcia SM, Spector E, Kronquist K, Creadon-Swindell G, Walter M, Christensen E, Van Hove JLK, Sass JO. D-Glyceric aciduria does not cause nonketotic hyperglycinemia: a historic co-occurrence. Mol Genet Metab. 2017;121:80–2. [PubMed: 28462797]
- Takayanagi M, Kure S, Sakata Y, Kurihara Y, Ohya Y, Kajita M, Tada K, Matsubara Y, Narisawa K. Human glycine decarboxylase gene (GLDC) and its highly conserved processed pseudogene (psiGLDC): their structure and expression, and the identification of a large deletion in a family with nonketotic hyperglycinemia. Hum Genet. 2000;106:298–305. [PubMed: 10798358]
- Tekgul H, Serdarolu G, Karapinar B, Plat M, Yurtsever S, Tosun A, Coker M, Gokben S. Vigabatrin caused rapidly progressive deterioration in two cases with early myoclonic encephalopathy associated with nonketotic hyperglycinemia. J Child Neurol. 2006;21:82–4. [PubMed: 16551461]
- Toone JR, Applegarth DA, Coulter-Mackie MB, James ER. Biochemical and molecular investigations of patients with nonketotic hyperglycinemia. Mol Genet Metab. 2000;70:116–21. [PubMed: 10873393]
- Toone JR, Applegarth DA, Coulter-Mackie MB, James ER. Recurrent mutations in P- and T-proteins of the glycine cleavage complex and a novel T-protein mutation (N145I): a strategy for the molecular investigation of patients with nonketotic hyperglycinemia (NKH). Mol Genet Metab. 2001;72:322–5. [PubMed: 11286506]
- Toone JR, Applegarth DA, Levy HL, Coulter-Mackie MB, Lee G. Molecular genetic and potential biochemical characteristics of patients with T-protein deficiency as a cause of glycine encephalopathy (NKH). Mol Genet Metab. 2003;79:272–80. [PubMed: 12948742]
- Tsao CY. The efficacy of vagus nerve stimulation in intractable epilepsy associated with nonketotic hyperglycinemia in two children. J Child Neurol. 2010;25:375–8. [PubMed: 19841478]
- van der Knaap MS, Wevers RA, Kure S, Gabreeëls FJ, Verhoeven NM, van Raaij-Selten B, Jaeken J. Increased cerebrospinal fluid glycine: a biochemical marker for a leukoencephalopathy with vanishing white matter. J Child Neurol. 1999;14:728–31. [PubMed: 10593550]
- Van Hove JL, Kishnani PS, Demaerel P, Kahler SG, Miller C, Jaeken J, Rutledge SL. Acute hydrocephalus in nonketotic hyperglycinemia. Neurology. 2000;54:754–6. [PubMed: 10680820]
- Van Hove JL, Vande Kerckhove K, Hennermann JB, Mahieu V, Declercq P, Mertens S, De Becker M, Kishnani PS, Jaeken J. Benzoate treatment and the glycine index in nonketotic hyperglycinaemia. J Inherit Metab Dis. 2005;28:651–63. [PubMed: 16151895]
- Wilson MP, Plecko B, Mills PB, Clayton PT. Disorders affecting vitamin B6 metabolism. J Inherit Metab Dis. 2019;42:629–46. [PubMed: 30671974]
- Wiltshire EJ, Poplawski NK, Harrison JR, Fletcher JM. Treatment of late-onset nonketotic hyperglycinaemia: effectiveness of imipramine and benzoate. J Inherit Metab Dis. 2000;23:15–21. [PubMed: 10682304]
- Yu TW, Chahrour MH, Coulter ME, Jiralerspong S, Okamura-Ikeda K, Ataman B, Schmitz-Abe K, Harmin DA, Adli M, Malik AN, D'Gama AM, Lim ET, Sanders SJ, Mochida GH, Partlow JN, Sunu CM, Felie JM, Rodriguez J, Nasir RH, Ware J, Joseph RM, Hill RS, Kwan BY, Al-Saffar M, Mukaddes NM, Hashmi A, Balkhy S, Gascon GG, Hisama FM, LeClair E, Poduri A, Oner O, Al-Saad S, Al-Awadi SA, Bastaki L, Ben-Omran T, Teebi AS, Al-Gazali L, Eapen V, Stevens CR, Rappaport L, Gabriel SB, Markianos K, State MW, Greenberg ME, Taniguchi H, Braverman NE, Morrow EM, Walsh CA. Using whole exome sequencing to identify inherited causes of autism. Neuron. 2013;77:259–73. [PMC free article: PMC3694430] [PubMed: 23352163]
Publication Details
Author Information and Affiliations
Section of Clinical Genetics and Metabolism
University of Colorado School of Medicine
Denver, Colorado
Section of Clinical Genetics and Metabolism
University of Colorado School of Medicine
Denver, Colorado
Section of Clinical Genetics and Metabolism
University of Colorado School of Medicine
Denver, Colorado
University Medical Center Mainz
Mainz, Germany
Publication History
Initial Posting: November 14, 2002; Last Update: May 23, 2019.
Copyright
GeneReviews® chapters are owned by the University of Washington. Permission is hereby granted to reproduce, distribute, and translate copies of content materials for noncommercial research purposes only, provided that (i) credit for source (http://www.genereviews.org/) and copyright (© 1993-2024 University of Washington) are included with each copy; (ii) a link to the original material is provided whenever the material is published elsewhere on the Web; and (iii) reproducers, distributors, and/or translators comply with the GeneReviews® Copyright Notice and Usage Disclaimer. No further modifications are allowed. For clarity, excerpts of GeneReviews chapters for use in lab reports and clinic notes are a permitted use.
For more information, see the GeneReviews® Copyright Notice and Usage Disclaimer.
For questions regarding permissions or whether a specified use is allowed, contact: ude.wu@tssamda.
Publisher
University of Washington, Seattle, Seattle (WA)
NLM Citation
Van Hove JLK, Coughlin C II, Swanson M, et al. Nonketotic Hyperglycinemia. 2002 Nov 14 [Updated 2019 May 23]. In: Adam MP, Feldman J, Mirzaa GM, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2024.

