Summary
Clinical characteristics.
PORCN-related developmental disorders include a spectrum of highly variable multisystem disorders caused by developmental abnormalities in mesodermal and ectodermal structures primarily involving the skin, limbs, eyes, and face. The manifestations vary among affected individuals, and many have only a subset of the characteristic features. Skin manifestations present at birth include atrophic and hypoplastic areas of skin; cutis aplasia; fat nodules in the dermis manifesting as soft, yellow-pink cutaneous nodules; and pigmentary changes. Verrucous papillomas of the skin and mucous membranes may appear later. The nails can be ridged, dysplastic, or hypoplastic; hair can be sparse or absent. Limb malformations include oligo- and syndactyly and split hand/foot. Developmental abnormalities of the eye can include anophthalmia/microphthalmia, iris and chorioretinal coloboma, and lacrimal duct abnormalities. Craniofacial findings can include facial asymmetry, notched alae nasi, cleft lip and palate, pointed chin, and small, underfolded pinnae. Dental anomalies can include hypodontia, enamel defects, and/or abnormally shaped teeth. Occasional findings include abdominal wall defects, diaphragmatic hernia, and renal anomalies. Psychomotor development is usually normal; some individuals have cognitive impairment and/or behavioral issues.
Diagnosis/testing.
A PORCN-related developmental disorder can be diagnosed in a proband with characteristic clinical features and a heterozygous pathogenic variant in PORCN identified by molecular genetic testing.
Management.
Treatment of manifestations: Care by a dermatologist for painful and pruritic erosive lesions that are prone to infection; laser therapy for atrophic areas and granulation tissue may be helpful; lotion for pruritic lesions; preoperative evaluation by an otolaryngologist for hypopharyngeal and/or tonsillar papillomas; management of large papillomas of the larynx, trachea, and/or esophagus per otolaryngologist or gastroenterologist; referral to a physical/occupational therapist and hand surgeon for management of hand and foot malformations; standard management of structural abnormalities of the eyes, kidneys, diaphragmatic hernia, and abdominal wall defects; visual aids or other resources for reduced vision; dental care, good oral hygiene, dietary counseling; consider fissure sealants to reduce dental caries; orthodontic care as needed for malocclusion, veneers as needed; hearing aids and community hearing services as needed; developmental services and educational support; standard management for behavioral issues.
Surveillance: Annual evaluation with a dermatologist; monitor for gastroesophageal reflux disease, swallowing difficulties, and symptoms of obstructive sleep apnea at each visit; annual physical examination for scoliosis, particularly in individuals with costovertebral segmentation abnormalities; annual eye examinations; dental examination every six months; annual hearing evaluation or as needed; assess growth and body composition to determine need for nutritional intervention at each visit; annual screening for cognitive, emotional, behavioral, and adaptive issues.
Agents/circumstances to avoid: Prevent exposure to extreme heat in those with hypohidrosis.
Genetic counseling.
PORCN-related developmental disorders are inherited in an X-linked manner. Females (90% of affected individuals) are heterozygous or mosaic for a PORCN pathogenic variant; most live-born affected males (10% of affected individuals) are mosaic for a de novo PORCN pathogenic variant. It is presumed that most non-mosaic hemizygous males are not viable. Approximately 95% of females with PORCN-related developmental disorders have a de novo pathogenic variant; ~5% inherited the pathogenic variant from a parent. The risk that the PORCN pathogenic variant will be transmitted by an affected heterozygous female is 50%; however, because most male conceptuses with a PORCN pathogenic variant are presumed to be spontaneously aborted, at delivery the expected sex ratio of offspring is: 33% unaffected females; 33% affected females; 33% unaffected males. If the affected female is mosaic for a PORCN pathogenic variant, the risk to her female offspring of inheriting the pathogenic variant depends on the level of mosaicism in her germline and can be as high as 50%. Once the PORCN pathogenic variant has been identified in an affected family member, prenatal and preimplantation genetic testing are possible.
Diagnosis
Suggestive Findings
A PORCN-related developmental disorder should be considered in an individual with any combination of the following characteristic ectodermal and limb findings and other common clinical findings.
Characteristic ectodermal manifestations (see Figure 1):
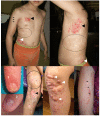
- Congenital patchy skin aplasia. Atrophic and hypoplastic areas of skin that often follow the lines of Blaschko and appear as depressed regions of pink or white color, often with a fibrous textureNote: The lines of Blaschko correspond to cell migration pathways evident during embryonic and fetal skin development. Like dermatomes, the lines of Blaschko are linear on the limbs and circumferential on the trunk. Unlike dermatomes, the lines of Blaschko do not correspond to innervation patterns.
- Congenital skin hypo- or hyperpigmentation often following a Blaschko linear distribution
- Telangiectasias. May be seen on the face, trunk, and extremities
- Congenital nodular fat herniation. Soft, yellow-pink nodules on the skin (which represent fat nodules in the dermis) typically seen on the trunk and extremities
- Congenital ridged, dysplastic, or hypoplastic nails
Characteristic limb malformations [Smith & Hunt 2016] (see Figure 2 and Figure 3):

Figure 2.
Hands showing syndactyly (black arrow) and split-hand malformation (black arrowhead) with only four digits (oligodactyly) on the left hand. The appearance of the left hand has been somewhat modified by partial surgical repair.
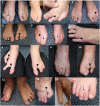
Figure 3.
Highly variable limb malformations Feet showing syndactyly (black arrows), split-foot malformation or ectrodactyly (black arrowheads), oligodactyly (3, 6, 7, 8, 10, 11, 12), and distal transverse limb defect (13)
- Syndactyly occurring variably on one or more extremities
- Ectrodactyly. Split-hand/foot malformation that may occur on ≥1 extremity
- Oligodactyly. Absent digit(s) may be seen in one or both hands and/or feet. Central digits are more frequently involved.
- Long bone reduction defect. Hypoplasia or shortening of the long bones in one or more extremities
- Transverse limb defect. Congenital absence of the distal portion of an upper and/or lower limb (e.g., hand, wrist, forearm, elbow) with no distal remaining portions, including acheiria or hemimelia
Other common clinical findings
- Hair. Hair shaft abnormalities on scanning electron microscopy, patchy alopecia of the scalp, wiry hair
- Verrucous papillomas of the skin and mucous membranes (including in the mouth, nose, larynx, esophagus, vaginal mucosa, and/or rectal mucosa)
- Dental abnormalities. Hypodontia, enamel defects / longitudinal grooving, peg teeth
- Other skin manifestations. Pebbled skin texture, photosensitivity
- Ocular manifestations. Iris colobomas, chorioretinal colobomas, microphthalmia, anophthalmia, cataracts, nystagmus, strabismus
Establishing the Diagnosis
Female proband. The diagnosis of a PORCN-related developmental disorder is established in a female proband with suggestive findings and a heterozygous pathogenic (or likely pathogenic) variant in PORCN identified by molecular genetic testing (see Table 1).
Male proband. The diagnosis of a PORCN-related developmental disorder is established in a male proband with suggestive findings and a hemizygous pathogenic (or likely pathogenic) variant in PORCN identified by molecular genetic testing (see Table 1). Note: Most affected males to date have somatic mosaicism for a hemizygous PORCN pathogenic variant [Lombardi et al 2011], although there are some recent reports of non-mosaic males with hypomorphic PORCN variants inherited from unaffected mothers [Happle 2021].
Note: (1) Per ACMG/AMG variant interpretation guidelines, the terms "pathogenic variant" and "likely pathogenic variant" are synonymous in a clinical setting, meaning that both are considered diagnostic, and both can be used for clinical decision making [Richards et al 2015]. Reference to "pathogenic variant" in this section is understood to include any likely pathogenic variant. (2) Identification of a heterozygous or hemizygous PORCN variant of uncertain significance does not establish or rule out the diagnosis.
Tiered molecular testing approaches can include a combination of single-gene testing (sequence analysis, gene-targeted deletion/duplication analysis), chromosomal microarray analysis (CMA), use of a multigene panel, and more comprehensive genomic testing.
- First-tier testing. On a blood sample, perform sequence analysis of PORCN. If no pathogenic variant is identified, perform either CMA (if not already performed) to detect large deletions/duplications that include PORCN or gene-targeted deletion/duplication analysis of PORCN if CMA was normal; note, however, that intragenic deletions detected by this method have been rarely reported (see Table 1).Note: (1) Several females and most males have somatic mosaicism for either a PORCN pathogenic variant or PORCN deletion; therefore, possible mosaicism must be considered when performing sequence analysis (see Second-tier testing). (2) A male with a 47,XXY karyotype and a heterozygous PORCN pathogenic variant on one of the two X chromosomes has been reported [Alkindi et al 2013].
- Second-tier testing. If first-tier testing does not detect a PORCN pathogenic variant or deletion, perform sequence analysis and deletion/duplication analysis on saliva or affected tissues (e.g., skin, papillomas, surgical specimens), which increase the sensitivity for detecting somatic mosaicism [Maas et al 2009].
- Testing to consider. If first-tier and second-tier testing do not detect a PORCN pathogenic variant or deletion, the following may also be considered:
- A multigene panel that includes PORCN and other genes of interest (see Differential Diagnosis). Note: (1) The genes included in the panel and the diagnostic sensitivity of the testing used for each gene vary by laboratory and are likely to change over time. (2) Some multigene panels may include genes not associated with the condition discussed in this GeneReview; thus, clinicians need to determine which multigene panel is most likely to identify the genetic cause of the condition while limiting identification of variants of uncertain significance and pathogenic variants in genes that do not explain the underlying phenotype. (3) In some laboratories, panel options may include a custom laboratory-designed panel and/or custom phenotype-focused exome analysis that includes genes specified by the clinician. (4) Methods used in a panel may include sequence analysis, deletion/duplication analysis, and/or other non-sequencing-based tests.
- Comprehensive genomic testing does not require the clinician to determine which gene is likely involved. Exome sequencing is most commonly used; genome sequencing is also possible.
Table 1.
Molecular Genetic Testing Used in PORCN-Related Developmental Disorders
Clinical Characteristics
Clinical Description
PORCN-related developmental disorders include a spectrum of highly variable multisystem disorders caused by developmental abnormalities in mesodermal and ectodermal structures primarily involving the skin, limbs, eyes, and face. The manifestations vary among affected individuals, and many have only a subset of the characteristic features.
Females account for 90% of individuals with PORCN-related developmental disorders.
The phenotypes in both males and females are highly variable due to tissue mosaicism: females have random X-chromosome inactivation (functional mosaicism), while most males have postzygotic somatic mosaicism.
Table 2.
PORCN-Related Developmental Disorders: Frequency of Select Features
Affected Females
Ectodermal manifestations. The most characteristic features of PORCN-related developmental disorders are the skin manifestations (see Figure 1). The cutaneous findings typically follow the lines of Blaschko and include patchy areas of skin aplasia/hypoplasia, skin hypo- and/or hyperpigmentation, and nodular fat herniation. The lines of Blaschko represent cell migration pathways that are linear on the limbs and circumferential on the trunk. The lines are classically described as V shaped overlying the upper spine, S shaped on the abdomen, and an inverted-U shape from the breast area to the upper arms. These findings are typically evident at birth, but the distribution and severity may change over time. Unilateral involvement has also been reported [Bree et al 2016].
Other integumentary system abnormalities include wiry hair, sparse hair, patchy alopecia of the scalp, and abnormal nails. The nails can be absent (anonychia), small (micronychia), hypoplastic, or dysplastic, often with longitudinal ridging, splitting, or V-shaped nicking [Bree et al 2016].
Papillomatosis. Papillomas and telangiectasias are typically not present at birth but develop with age.
- Verrucous papillomas are found in the oral mucosa of the mouth, nose, pharynx, larynx, trachea, and esophagus. Large papillomas of the larynx can obstruct breathing during anesthesia or can cause obstructive sleep apnea. Papillomas in the esophagus or larynx can also cause or contribute to severe gastroesophageal reflux disease (GERD).
- The vaginal or rectal mucosa are also common sites for papillomas, where they can be confused with genital warts.
Dental abnormalities and eye findings, both of which result from abnormalities in ectodermal appendage development, are described separately below.
Limb and skeletal manifestations. Most individuals with a PORCN-related developmental disorder have limb malformations noted at birth, including syndactyly, oligodactyly, and split-hand/foot malformation or ectrodactyly (see Figures 2 and 3). These malformations, which do not change over time, may impair function.
Additionally, reduction defects of the long bones ranging from leg length discrepancies to transverse defects of the distal radius/ulna or tibia/fibula are commonly seen.
Less common limb malformations that may be present at birth and impair function include camptodactyly (contraction deformities of the digits) and brachydactyly (shortening of the digits).
Costovertebral segmentation abnormalities including fused ribs, bifid ribs, hemivertebrae, and butterfly vertebrae are present at birth but are often not evident on physical examination and may only be seen on x-ray of the chest and/or spine. Although these malformations do not typically cause problems in infancy or early childhood, they may cause scoliosis as the child grows. Kyphosis or kyphoscoliosis is seen in approximately 10% of affected individuals [Smith & Hunt 2016]. More often, these segmentation abnormalities do not cause health issues.
Diastasis pubis, an abnormal separation of the symphysis pubis, may be an incidental finding or may present in adolescence or adulthood with pain. The gap between the pubic bones in the average non-pregnant adult is 4-5 mm. An abnormal gap is considered to be 1 cm or more, sometimes with the two bones being slightly out of alignment. In some individuals, diastasis pubis may cause pain with walking or in the symphysis pubis, legs, groin, and lower abdomen.
Fibrous dysplasia of bone (i.e., replacement of medullary bone with trabeculae of woven bone containing fluid-filled cysts embedded in a fibrous matrix) may affect any bone at any time. On x-ray the bone appears radiolucent, with what is classically described as a "ground-glass" appearance. Fibrous dysplasia may be asymptomatic or become evident when it is the site of a pathologic fracture.
Giant cell-like tumors of long bones, reported on occasion, may develop in childhood, adolescence, or adulthood. They typically become evident when a pathologic bone fracture occurs at the site of the lesion [Selzer et al 1974, Joannides et al 1983, Tanaka et al 1990]. In the small number of reports to date, none of these tumors has been malignant.
Osteopathia striata, a striated appearance of the bones evident on plain x-rays, is common and may be seen in childhood, adolescence, and adulthood. It is currently unclear if individuals with this finding are at increased risk for general osteoporosis. Of note, a spontaneous patella fracture related to osteoporosis in an individual with a PORCN-related developmental disorder has been reported [Altschuler et al 2012].
Eye findings. Developmental abnormalities of the eyes are common and are evident at birth; PORCN plays an important role in the development of the optic cup [Fuhrmann et al 2022]. Depending on the severity of the manifestations, vision can range from 20/20 to no light perception. Reported eye abnormalities include anophthalmia/microphthalmia; microcornea; iris, chorioretinal, and eyelid colobomas; lacrimal duct abnormalities; and cataracts (cortical and subcapsular) [Gisseman & Herce 2016].
Strabismus and/or nystagmus can be observed when visual impairment in infancy is significant.
Craniofacial findings. Facial features are variable and include facial asymmetry, notched alae nasi, pointed chin, and small, underfolded pinnae. These facial characteristics are not typically evident at birth but develop with time (see Figure 4) [Bostwick et al 2016].

Figure 4.
Note facial features of pointed chin and small right ear.
Cleft lip and palate can be present and may lead to difficulty with feeding. More severe facial clefting can cause feeding, breathing, and vision problems, as well as significant cosmetic concerns [Wright et al 2016].
Oral and dental findings. Oral manifestations are seen in more than half of affected individuals and include both soft tissue and hard tissue abnormalities.
Enamel hypoplasia that predisposes to dental caries is the most common problem. Other findings include: hypodontia, oligodontia, supernumerary teeth, and dental crowding leading to malocclusion of both primary and secondary dentition; vertical grooving of the teeth; microdontia (small teeth); taurodontia (prism-shaped molars); fused teeth; and abnormal root morphology [Balmer et al 2004, Tejani et al 2005, Murakami et al 2011]. Affected individuals may also have problems with the eruption and position of teeth.
Soft tissue abnormalities include generalized gingivitis and intraoral lipomas and papillomas [Wright et al 2016].
Gastrointestinal and nutrition. Findings include poor weight gain (77% of individuals), short stature (65%), oral motor dysfunction (41%),GERD (24%), gastroparesis (35%), and constipation (35%) [Motil et al 2016, Hsu et al 2019]. Food allergies – primarily to milk, soy, and shellfish – are present in 12% of affected individuals.
Other developmental abnormalities of the digestive system are rare but may have severe consequences; they include abdominal wall defects and diaphragmatic hernia (see Congenital Diaphragmatic Hernia Overview).
Severe GERD has been reported in infancy and childhood, leading to feeding difficulties with frequent vomiting and/or discomfort/distress. GERD likely results from esophageal papillomas [Brinson et al 1987].
Renal and urogenital. Genital labial hypoplasia is present in most females [Adeyemi-Fowode et al 2016]. Occasional affected individuals with müllerian anomalies, including bicornuate uterus, have been described [Reddy & Laufer 2009, Lopez-Porras et al 2011]. Renal structural abnormalities are uncommon.
Cognitive and psychological. Development and intellectual ability are normal in most individuals. Intellectual impairment (15%-20% of individuals), behavioral issues (~20%), emotional lability (40%-50%), and withdrawn behavior (65%) have been reported [Deidrick et al 2016]. In those with emotional, behavioral, adaptive, and intellectual impairment, the spectrum of severity varies widely.
Structural brain abnormalities and spina bifida [Goltz et al 1970, Almeida et al 1988] have been reported but are uncommon.
Epilepsy has been reported [Kanemura et al 2011].
Other. Mixed conductive and sensorineural hearing loss has been reported on occasion.
An adult female with multiple cutaneous basal cell carcinomas has been reported. Whether basal cell carcinoma is more prevalent in individuals with a PORCN-related developmental disorder is currently unknown, but heightened surveillance and appropriate treatment for such lesions may be indicated [Patrizi et al 2012].
When the first affected female in the family has milder manifestations than affected females in subsequent generations [Heinz et al 2019], it is most likely that she has either mosaicism for the PORCN pathogenic variant or skewing of X-chromosome inactivation. Alternative explanations could be reduced reproductive fitness in severely affected females, such that only mildly affected females reproduce.
Affected Males
Because relatively few affected males have been reported, no comprehensive data for a "typical" male phenotype exist.
Affected males may have any of the features seen in affected females, including typical skin findings; sparse, brittle hair; nail dystrophy; microphthalmia; syndactyly; split-hand/foot malformation; costovertebral segmentation abnormalities; osteopathia striata; and diastasis pubis [Wang et al 2007, Bornholdt et al 2009, Maas et al 2009, Lasocki et al 2011, Lombardi et al 2011, Vreeburg et al 2011, Yoshihashi et al 2011].
Affected males most often have somatic mosaicism for a PORCN pathogenic variant and are generally more mildly affected than females [Grzeschik et al 2007, Wang et al 2007, Lombardi et al 2011]. Of note, fathers are typically more mildly affected than their daughters [Burgdorf et al 1981], a discrepancy attributed to mosaicism in the males.
Pathology
Histopathologic and ultrastructural studies of the skin have shown the following:
- A thinned dermis with disordered connective tissue and decreased number of collagen bundles and elastin fibers [Kanitakis et al 2003]
- Rests of mature adipose tissue scattered throughout the reticular and papillary dermis [Howell & Freeman 1989, del Carmen Boente et al 2007]. Whether these represent herniation of fat into a thinned dermis or ectopic aggregation of fat within a dysplastic dermis is unclear [Howell & Freeman 1989].
- Verrucous papillomas that resemble squamous papillomas with hyperplastic, stratified squamous epithelium overlying a fibrovascular core. Verrucous papillomas lack the typical morphologic evidence of human papilloma virus infection and stain negative for Epstein-Barr virus RNA [Rosen & Bocklage 2005].
- From biopsies from Blaschkoid streaks, findings of increased papillary dermal blood vessels, decreased thickness of the dermis, and adipocytes high in the dermis strongly point to the diagnosis of a PORCN-related developmental disorder [Ko et al 2016].
Genotype-Phenotype Correlations
Information on genotype-phenotype correlations in PORCN-related developmental disorders is limited.
- Available data suggest that the level of X-chromosome inactivation correlates with severity of the phenotype in some (familial) cases [Grzeschik et al 2007, Wang et al 2007].Note: All females with deletions in PORCN have extremely skewed X-chromosome inactivation, whereas females with a single-nucleotide variant can have random or skewed X-chromosome inactivation [Grzeschik et al 2007, Wang et al 2007, Lombardi et al 2011].
- Most affected males to date have somatic mosaicism for a PORCN pathogenic variant, with males being generally more mildly affected than females; however, some severely affected males have been reported [Maas et al 2009, Bornholdt et al 2009, Lombardi et al 2011].
- Non-mosaic PORCN variants have been identified in ten males from four families with isolated anophthalmia/microphthalmia or multiple congenital anomalies. Heterozygous female family members were asymptomatic or presented with mild features, suggesting a hypomorphic PORCN variant [Brady et al 2015, Madan et al 2017, Happle 2021, Wawrocka et al 2021].
Penetrance
PORCN-related developmental disorders are typically highly penetrant in females, but the phenotypic severity can occasionally be mitigated by skewed X-chromosome inactivation or presence of a hypomorphic variant.
Most males are mosaic for a hemizygous somatic PORCN pathogenic variant and can be so mildly affected as to not come to medical attention until adulthood.
Nomenclature
The title of this GeneReview – "PORCN-related developmental disorders" – is preferred by the author over the designation "focal dermal hypoplasia" because not all individuals with the disorder have focal areas of skin hypoplasia and because "PORCN-related developmental disorders" more accurately represents the highly variable multisystemic nature of the syndrome.
"Gorlin-Goltz syndrome" is another name for nevoid basal cell carcinoma syndrome.
Prevalence
PORCN-related developmental disorders are uncommon, with about 300 reported affected individuals worldwide [Goltz 1992, Tadini et al 2015]. The exact prevalence is unknown.
Genetically Related (Allelic) Disorders
No phenotypes other than those discussed in this GeneReview are known to be associated with pathogenic variants in PORCN.
Differential Diagnosis
Table 3.
Genes of Interest in the Differential Diagnosis of PORCN-Related Developmental Disorders
Oculocerebrocutaneous syndrome (OMIM 164180) – a disorder of unknown genetic cause – is characterized by microphthalmia/anophthalmia, orbital cysts, linear skin pigmentation, and dermal hypoplasia. The condition predominantly affects males and can be distinguished from PORCN-related developmental disorders by the presence (in oculocerebrocutaneous syndrome) of characteristic brain malformations including frontal polymicrogyria, periventricular nodular heterotopia, and agenesis of the corpus callosum [Moog et al 2005].
Other. Papillomas of the genital and anal region are common and should not be confused with genital warts.
Note. Individuals with PORCN-related developmental disorders have been diagnosed with angioma serpiginosum [Blinkenberg et al 2007, Houge et al 2008, Happle 2009].
Management
No clinical practice guidelines for PORCN-related developmental disorders have been published.
Evaluations Following Initial Diagnosis
To establish the extent of disease and needs in an individual diagnosed with a PORCN-related developmental disorder, the evaluations summarized in Table 4 (if not performed as part of the evaluation that led to the diagnosis) are recommended.
Table 4.
PORCN-Related Developmental Disorders: Recommended Evaluations Following Initial Diagnosis
Treatment of Manifestations
Supportive care to improve quality of life, maximize function, and reduce complications is recommended. This ideally involves multidisciplinary care by specialists in relevant fields (see Table 5).
Table 5.
PORCN-Related Developmental Disorders: Treatment of Manifestations
Surveillance
To monitor existing manifestations, the individual's response to supportive care, and the emergence of new manifestations, the evaluations summarized in Table 6 are recommended.
Table 6.
PORCN-Related Developmental Disorders: Recommended Surveillance
Agents/Circumstances to Avoid
Because some individuals with severe skin manifestations may have hypohidrosis (and thus be at increased risk for heat intolerance), care should be taken to prevent exposure to extreme heat.
Evaluation of Relatives at Risk
See Genetic Counseling for issues related to testing of at-risk relatives for genetic counseling purposes.
Pregnancy Management
For affected women, management of pregnancy should be guided by standard obstetric principles, taking into account potential complications of PORCN-related developmental disorders. Skeletal abnormalities including scoliosis or diastasis pubis may be present in some affected women and may affect delivery management. Women with significant scoliosis will benefit from evaluation of respiratory status and feasibility of epidural analgesia. Obstetricians should be aware that verrucous papillomas in genital areas in women with a PORCN-related developmental disorder are unlikely to be viral in origin, and thus there is no risk for transmission to the newborn during vaginal delivery.
Therapies Under Investigation
Search ClinicalTrials.gov in the US and EU Clinical Trials Register in Europe for access to information on clinical studies for a wide range of diseases and conditions. Note: There may not be clinical trials for this disorder.
Genetic Counseling
Genetic counseling is the process of providing individuals and families with information on the nature, mode(s) of inheritance, and implications of genetic disorders to help them make informed medical and personal decisions. The following section deals with genetic risk assessment and the use of family history and genetic testing to clarify genetic status for family members; it is not meant to address all personal, cultural, or ethical issues that may arise or to substitute for consultation with a genetics professional. —ED.
Mode of Inheritance
PORCN-related developmental disorders are inherited in an X-linked manner.
Females account for 90% of individuals with PORCN-related developmental disorders; they may have heterozygous or mosaic pathogenic variants in PORCN. Males account for 10% of individuals with PORCN-related developmental disorders; nearly all live-born affected males who have had molecular genetic testing are mosaic for a PORCN pathogenic variant [Lombardi et al 2011]. It is presumed that most non-mosaic, hemizygous males are not viable.
Risk to Family Members
Parents of a female proband
- Approximately 95% of females with a PORCN-related developmental disorder represent simplex cases (i.e., the only family member known to be affected) and have a PORCN-related developmental disorder as the result of a either a de novo germline pathogenic variant, a somatic mosaic pathogenic variant, or a pathogenic variant inherited from an unaffected parent with low-level mosaicism.
- Approximately 5% of females with a PORCN-related developmental disorder have inherited a PORCN pathogenic variant from an affected parent, usually the mother.
- Female probands with a maternally inherited PORCN pathogenic variant may be more severely affected than their mothers [Shimaoka et al 2009, Yesodharan et al 2018]. When the mother of the proband is the first affected female in the family and has milder manifestations than affected females in subsequent generations [Wechsler et al 1988, Kilmer et al 1993], it is most likely that the mother has either mosaicism for the PORCN pathogenic variant or skewing of X-chromosome inactivation.
- Less frequently, an affected female inherited the pathogenic variant from a father mosaic for a PORCN pathogenic variant [Wang et al 2007]; fathers with a PORCN-related developmental disorder are typically more mildly affected than their daughters [Burgdorf et al 1981].
- Detailed evaluation of the parents and review of the extended family history may help distinguish probands with a de novo pathogenic variant from those with an inherited pathogenic variant. If a germline PORCN pathogenic variant has been identified in the proband, * molecular genetic testing of the parent who has manifestations of a PORCN-related developmental disorder is appropriate. If neither parent has clinical manifestations of a PORCN-related developmental disorder, molecular genetic testing of both parents should be considered because: (1) the father or mother may have low-level mosaicism or (2) the mother may be a mildly affected (or apparently asymptomatic) heterozygote secondary to extremely favorable skewing of X-chromosome inactivation and/or the class of pathogenic variant segregating in the family [Brady et al 2015, Happle 2021].* If a proband with a PORCN-related developmental disorder has the disorder as the result of a somatic mosaic PORCN pathogenic variant (i.e., a variant resulting from mutation that occurs in the proband during embryonic development), the parents do not have the pathogenic variant.
- If the proband has a germline PORCN pathogenic variant that is not identified in either parent and parental identity testing has confirmed biological maternity and paternity, the following possibilities should be considered:
- The proband has a de novo germline pathogenic variant.
- The proband inherited a pathogenic variant from a mosaic parent (a parent with somatic and germline mosaicism may be apparently asymptomatic or mildly/minimally affected).Note: Testing parents using peripheral blood leukocyte-derived DNA may not detect all instances of somatic mosaicism. Molecular genetic tests sensitive enough to detect low-level somatic mosaicism, such as high-coverage next-generation sequencing or allele-specific PCR, should therefore be considered. Testing of parental leukocyte DNA will not detect a pathogenic variant that is present in the germ cells only.
- An apparently negative family history cannot be confirmed until appropriate evaluations have been performed.
Parents of a male proband
- Live-born affected males are rare, and most affected males have somatic mosaicism for a de novo, presumably postzygotic pathogenic variant. The mother of a male proband with a somatic mosaic postzygotic PORCN pathogenic variant does not have the pathogenic variant.
- If a germline PORCN pathogenic variant has been identified in an affected male, the mother should be examined for subtle features of a PORCN-related developmental disorder, and maternal molecular genetic testing for the PORCN pathogenic variant identified in the proband is indicated. Hemizygous affected males with a range of clinical findings have been reported as offspring of seemingly asymptomatic mothers with a presumed hypomorphic pathogenic variant and, in some, favorable skewing of X-chromosome inactivation [Brady et al 2015, Happle 2021] (see Genotype-Phenotype Correlations).
- The father of an affected male will not have the disorder nor will he be hemizygous for the PORCN pathogenic variant; therefore, he does not require further evaluation/testing.
Sibs of a female proband. The risk to sibs of a female proband depends on the genetic status of the mother and father:
- If the mother of the proband:
- Is affected and/or known to be heterozygous for a PORCN pathogenic variant, the risk at conception of a sib inheriting the variant is 50%. However, the risk at delivery that a sib will be affected is lower than 50% because nearly all male conceptuses with the variant are presumed to be spontaneously aborted. The expected ratio in live-born sibs is 33% unaffected females, 33% affected females, and 33% unaffected males.
- Is mosaic for a PORCN pathogenic variant, the risk at conception of a sib inheriting the pathogenic variant is as high as 50% depending on the level of mosaicism in the mother's germline [Heinz et al 2019].
- Is clinically unaffected and a PORCN pathogenic variant identified in the proband cannot be detected in maternal leukocyte DNA, the risk to sibs is greater than that of the general population because of the possibility of low-level parental mosaicism.
- Is clinically unaffected but her genetic status is unknown, the risk to sibs is greater than that of the general population because of the possibility of low-level parental mosaicism and the possibility that the mother is heterozygous with favorable skewing of X-chromosome inactivation or heterozygous for a hypomorphic PORCN pathogenic variant [Brady et al 2015, Happle 2021] (see Genotype-Phenotype Correlations).
- If the father has the PORCN pathogenic variant, the risk to female sibs of inheriting the variant at conception is as high as 100% depending on the level of mosaicism in the father's germline. Male sibs are not at risk of inheriting the pathogenic variant from their father.
Sibs of a male proband. The risk to sibs depends on the genetic status of the mother:
- If a male proband is mosaic for a postzygotic pathogenic variant (most live-born males are mosaic based on evidence from both molecular studies and clinical reports), the mother does not have the PORCN pathogenic variant and the risk to the sib of an affected male is similar to the population risk for PORCN-related developmental disorders.
- If the mother of the proband has a PORCN pathogenic variant, the chance of the mother transmitting it in each pregnancy is 50%. Although most male conceptuses with a PORCN pathogenic variant are presumed to be spontaneously aborted, transmission of PORCN pathogenic variants to affected, hemizygous males from seemingly asymptomatic mothers with a presumed hypomorphic variant and, in some, favorable skewing of X-chromosome inactivation has been reported [Brady et al 2015, Happle 2021].
Offspring of female proband. The risk to the offspring of females with a PORCN-related developmental disorder must take into consideration the presumed lethality to males during gestation.
- At conception, the risk that the PORCN pathogenic variant will be transmitted is 50%; however, most male conceptuses with the PORCN variant are presumed to be spontaneously aborted. Thus, at delivery the expected ratio of offspring is 33% unaffected females, 33% affected females, and 33% unaffected males.
- If the proband is mosaic for a PORCN pathogenic variant, the risk to her offspring is as high as 50%, depending on the level of mosaicism in her germline [Heinz et al 2019].
Offspring of male proband. Most males with a PORCN-related developmental disorder have somatic mosaicism for a PORCN pathogenic variant.
- The risk to an affected male of having an affected daughter is as high as 100% depending on the level of mosaicism in his germline. The daughter of a male proband will typically be more severely affected than the male proband [Burgdorf et al 1981].
- Males do not transmit their X chromosome to their sons and thus their sons are not at risk of inheriting the PORCN pathogenic variant.
Other family members. If the mother of the proband also has a PORCN pathogenic variant, her female family members may also be at risk of having the pathogenic variant (asymptomatic or symptomatic) and her father may be at risk of being mosaic for the pathogenic variant.
Related Genetic Counseling Issues
Family planning
- The optimal time for determination of genetic risk and discussion of the availability of prenatal/preimplantation genetic testing is before pregnancy.
- It is appropriate to offer genetic counseling (including discussion of potential risks to offspring and reproductive options) to affected individuals and apparently asymptomatic female family members at risk of having a PORCN pathogenic variant.
Prenatal Testing and Preimplantation Genetic Testing
Molecular genetic testing. Once the PORCN pathogenic variant has been identified in an affected family member, prenatal and preimplantation genetic testing for a PORCN-related developmental disorder are possible.
Ultrasound examination. The combination of intrauterine growth restriction or low average weight, limb malformation, and thoraco-abdominal wall defect or diaphragmatic hernia was a consistent finding in 11 fetuses found to have PORCN pathogenic variants [Mary et al 2017]. Identification of this combination of features on routine prenatal ultrasound examination may raise the possibility of a PORCN-related developmental disorder in a fetus not known to be at increased risk.
Differences in perspective may exist among medical professionals and within families regarding the use of prenatal testing. While most centers would consider use of prenatal testing to be a personal decision, discussion of these issues may be helpful.
Resources
GeneReviews staff has selected the following disease-specific and/or umbrella support organizations and/or registries for the benefit of individuals with this disorder and their families. GeneReviews is not responsible for the information provided by other organizations. For information on selection criteria, click here.
- Ectodermal Dysplasia SocietyUnited KingdomPhone: 01242 261332Email: [email protected]
- National Foundation for Ectodermal Dysplasias (NFED)Phone: 618-566-2020Email: [email protected]
- Ectodermal Dysplasias International RegistryEmail: [email protected]
Molecular Genetics
Information in the Molecular Genetics and OMIM tables may differ from that elsewhere in the GeneReview: tables may contain more recent information. —ED.
Table A.
PORCN-Related Developmental Disorders: Genes and Databases
Table B.
OMIM Entries for PORCN-Related Developmental Disorders (View All in OMIM)
Molecular Pathogenesis
PORCN encodes protein-serine O-palmitoleoyltransferase porcupine (PORCN), a protein expressed in a wide variety of tissues that palmitoylates various Wnt proteins. In model organisms, the palmitoylation of Wnt proteins by PORCN has been shown to be required for secretion and signaling of most Wnt proteins from Wnt-producing cells [van Amerongen & Nusse 2009, Chen et al 2012, Clevers & Nusse 2012] and may play a role in Wnt protein levels. Wnt signaling is required for induction, proliferation, morphogenesis, and maintenance of most organs. Wnt proteins are important secreted morphogens that interact with receptors and coreceptors on target cells. Activation of the Wnt pathway is important for normal development [Clevers & Nusse 2012] and may be required to activate additional non-canonical Wnt signaling [Proffitt & Virshup 2012]. Wnt-3a is retained in the endoplasmic reticulum of cultured cells when Porcn is inactivated [Takada et al 2006, Clevers & Nusse 2012]. Studies of skin fibroblasts from females with a PORCN-related developmental disorder have demonstrated that PORCN activation of Wnt proteins is required for cellular reprograming [Ross et al 2014].
Loss of function of orthologs in mouse cells and Drosophila results in failure of Wnt proteins to be secreted from the endoplasmic reticulum in Wnt-producing cells, with defective downstream Wnt signaling [Tanaka et al 2000, Takada et al 2006]. Inactivation of Porcn in mouse embryos has resulted in early embryonic lethality and revealed that it is required for gastrulation and normal development of mesoderm- and ectoderm-derived structures [Barrott et al 2011, Biechele et al 2011, Liu et al 2012]. Conditional inactivation of Porcn in developing skin causes alopecia because hair follicles do not form [Liu et al 2012], and in developing limbs causes skeletal defects reminiscent of those seen in persons with a PORCN-related developmental disorder [Barrott et al 2011, Liu et al 2012].
Mechanism of disease causation. Loss of function
PORCN-specific laboratory technical considerations. To date most affected males have somatic mosaicism for a PORCN pathogenic variant [Lombardi et al 2011].
Chapter Notes
Author Notes
Author History
Bret Bostwick, MD; Baylor College of Medicine Houston (2016-2023)
Ignatia B Van den Veyver, MD; Baylor College of Medicine Houston (2008-2023)
V Reid Sutton, MD (2008-present)
Revision History
- 15 June 2023 (sw) Comprehensive update posted live
- 21 July 2016 (bp) Comprehensive update posted live
- 11 April 2013 (me) Comprehensive update posted live
- 15 May 2008 (me) Review posted live
- 6 February 2008 (vrs) Original submission
References
Literature Cited
- Adeyemi-Fowode OA, Mansouri R, Dietrich JE. Gynecologic findings in Goltz syndrome: a case series. Am J Med Genet C Semin Med Genet. 2016;172C:64–6. [PubMed: 27001927]
- Alkindi S, Battin M, Aftimos S, Purvis D. Focal dermal hypoplasia due to a novel pathogenic variant in a boy with Klinefelter syndrome. Pediatr Dermatol. 2013;30:476–9. [PubMed: 23131169]
- Almeida L, Anyane-Yeboa K, Grossman M, Rosen T. Myelomeningocele, Arnold-Chiari anomaly and hydrocephalus in focal dermal hypoplasia. Am J Med Genet. 1988;30:917–23. [PubMed: 3189414]
- Altschuler EL, Yoon RS, Dentico R, Liporace FA. Spontaneous patella fracture presenting as osteomyelitis in focal dermal hypoplasia. Knee. 2012;19:500–3. [PubMed: 22000280]
- Balmer R, Cameron AC, Adès L, Aldred MJ. Enamel defects and lyonization in focal dermal hypoplasia. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 2004;98:686–91. [PubMed: 15583541]
- Barrott JJ, Cash GM, Smith AP, Barrow JR, Murtaugh LC. Deletion of mouse Porcn blocks Wnt ligand secretion and reveals an ectodermal etiology of human focal dermal hypoplasia/Goltz syndrome. Proc Natl Acad Sci U S A. 2011;108:12752–7. [PMC free article: PMC3150921] [PubMed: 21768372]
- Bertani H, Mirante VG, Caruso A, Manno M, Brancaccio ML, Conigliaro R. Successful treatment of diffuse esophageal papillomatosis with balloon-assisted radiofrequency ablation in a patient with Goltz syndrome. Endoscopy. 2014;46:E404–5. [PubMed: 25314164]
- Biechele S, Cox BJ, Rossant J. Porcupine homolog is required for canonical Wnt signaling and gastrulation in mouse embryos. Dev Biol. 2011;355:275–85. [PubMed: 21554866]
- Blinkenberg EO, Brendehaug A, Sandvik AK, Vatne O, Hennekam RCM, Houge G. Angioma serpiginosum with oesophageal papillomatiosis is an X-linked dominant condition that maps to Xp11.3-Xq12. Eur J Hum Genet. 2007;15:543–7. [PubMed: 17342156]
- Bornholdt D, Oeffner F, König A, Happle R, Alanay Y, Ascherman J, Benke PJ, Boente Mdel C, van der Burgt I, Chassaing N, Ellis I, Francisco CR, Della Giovanna P, Hamel B, Has C, Heinelt K, Janecke A, Kastrup W, Loeys B, Lohrisch I, Marcelis C, Mehraein Y, Nicolas ME, Pagliarini D, Paradisi M, Patrizi A, Piccione M, Piza-Katzer H, Prager B, Prescott K, Strien J, Utine GE, Zeller MS, Grzeschik KH. PORCN mutations in focal dermal hypoplasia: coping with lethality. Hum Mutat. 2009;30:E618–28. [PubMed: 19309688]
- Bostwick B, Fang P, Patel A, Sutton VR. Phenotypic and molecular characterization of focal dermal hypoplasia in 18 individuals. Am J Med Genet C Semin Med Genet. 2016;172C:9–20. [PubMed: 26853229]
- Brady PD, Van Esch H, Fieremans N, Froyen G, Slavotinek A, Deprest J, Devriendt K, Vermeesch JR. Expanding the phenotypic spectrum of PORCN variants in two males with syndromic microphthalmia. Eur J Hum Genet. 2015;23:551–4. [PMC free article: PMC4666577] [PubMed: 25026905]
- Bree AF, Grange DK, Hicks MJ, Goltz RW. Dermatologic findings of focal dermal hypoplasia (Goltz syndrome). Am J Med Genet C Semin Med Genet. 2016;172C:44–51. [PubMed: 26858134]
- Brinson RR, Schuman BM, Mills LR, Thigpen S, Freedman S. Multiple squamous papillomas of the esophagus associated with Goltz syndrome. Am J Gastroenterol. 1987;82:1177–9. [PubMed: 3673998]
- Burgdorf WHC, Dick GF, Soderberg MD, Goltz RW. Focal dermal hypoplasia in a father and daughter. J Am Acad Dermatol. 1981;4:273–7. [PubMed: 7217396]
- Chen D, Li Y, Zhou Z, Xing Y, Zhong Y, Zou X, Tian W, Zhang C. Synergistic inhibition of Wnt pathway by HIF-1α and osteoblast-specific transcription factor osterix (Osx) in osteoblasts. PLoS One. 2012;7:e52948. [PMC free article: PMC3531395] [PubMed: 23300831]
- Clevers H, Nusse R. Wnt/beta-catenin signaling and disease. Cell. 2012;149:1192–205. [PubMed: 22682243]
- Deidrick KK, Early M, Constance J, Stein M, Fete TJ. Cognitive and psychological functioning in focal dermal hypoplasia. Am J Med Genet C Semin Med Genet. 2016;172C:34–40. [PubMed: 26818018]
- del Carmen Boente M, Asial RA, Winik BC. Focal dermal hypoplasia: ultrastructural abnormalities of the connective tissue. J Cutan Pathol. 2007;34:181–7. [PubMed: 17244031]
- Fernandes PH, Wen S, Sutton VR, Ward PA, Van den Veyver IB, Fang P. PORCN pathogenic variants and variants identified in patients with focal dermal hypoplasia through diagnostic gene sequencing. Genet Test Mol Biomarkers. 2010;14:709–13. [PubMed: 20854095]
- Fuhrmann S, Ramirez S, Mina Abouda M, Campbell CD. PORCN is essential for growth and invagination of the mammalian optic cup. Front Cell Dev Biol. 2022;10:1016182. [PMC free article: PMC9661423] [PubMed: 36393832]
- Gisseman JD, Herce HH. Ophthalmologic manifestations of focal dermal hypoplasia (Goltz syndrome): a case series of 18 patients. Am J Med Genet C Semin Med Genet. 2016;172C:59–63. [PubMed: 27001926]
- Goltz RW. Focal dermal hypoplasia syndrome. An update. Arch Dermatol. 1992;128:1108–11. [PubMed: 1497368]
- Goltz RW, Henderson RR, Hitch JM, Ott JE. Focal dermal hypoplasia syndrome. A review of the literature and report of two cases. Arch Dermatol. 1970;101:1–11. [PubMed: 5416790]
- Grzeschik KH, Bornjoldt D, Oeffner F, König A, del Carmen Boente M, Enders H, Hertl M, Grasshoff U, Höfling K, Oji V, Paradisi M, Schuchardt C, Szalai Z, Tadini G, Traupe H, Happle R. Deficiency of PORCN, a regulator of Wnt signaling, is associated with focal dermal hypoplasia. Nat Genet. 2007;39:833–5. [PubMed: 17546031]
- Happle R. Angioma serpiginosum is not caused by PORCN pathogenic variants. Eur J Hum Genet. 2009;17:881–2. [PMC free article: PMC2986492] [PubMed: 19337307]
- Happle R. The PORCN non-Goltz spectrum (PONGOS): a new group of genetic disorders. Am J Med Genet. 2021;185A:13–14. [PubMed: 33219742]
- Heinz L, Bourrat E, Vabres P, Thevenon J, Hotz A, Hörer S, Küsel J, Zimmer AD, Alter S, Happle R, Fischer J. Mosaicism due to postzygotic mutations in women with focal dermal hypoplasia. Br J Dermatol. 2019;180:657–61. [PubMed: 30022487]
- Houge G, Oeffner F, Grzeschik KH. An Xp11.23 deletion containing PORCN may also cause angioma serpiginosum, a cosmetic skin disease associated with extreme skewing of X-inactivation. Eur J Hum Genet. 2008;16:1027–8. [PubMed: 18478042]
- Howell JB, Freeman RG. Cutaneous defects of focal dermal hypoplasia: an ectomesodermal dysplasia syndrome. J Cutan Pathol. 1989;16:237–58. [PubMed: 2592623]
- Hsu SC, Bartz S, Pyle L, Fete M, Davis S, Ohman-Hanson R, Fete TJ, Motil KJ. Growth failure in focal dermal hypoplasia. Am J Med Genet. 2019;179A:628–633. [PMC free article: PMC6435029] [PubMed: 30693654]
- Joannides T, Pringle JAS, Shaw DG, Godlee JN, Kemp HB. Giant cell tumour of bone in focal dermal hypoplasia. Br J Radiol. 1983;56:684–5. [PubMed: 6883033]
- Kanemura H, Hatakeyama K, Sugita K, Aihara M. Epilepsy in a patient with focal dermal hypoplasia. Pediatric neurology. 2011;44:135–8. [PubMed: 21215914]
- Kanitakis J, Souillet AL, Butnaru C, Claudy A. Melanocyte stimulation in focal dermal hypoplasia with unusual pigmented skin lesions: a histologic and immunohistochemical study. Pediatr Dermatol. 2003;20:249–53. [PubMed: 12787276]
- Kashyap P, Sweetser S, Farrugia G. Esophageal papillomas and skin abnormalities. Focal dermal hypoplasia (Goltz syndrome) manifesting with esophageal papillomatosis. Gastroenterology. 2011;140:784. [PubMed: 21272558]
- Kilmer SL, Grix AW Jr, Isseroff RR. Focal dermal hypoplasia: four cases with widely varying presentations. J Am Acad Dermatol. 1993;28:839–43. [PubMed: 8491876]
- Ko CJ, Antaya RJ, Zubek A, Craiglow B, Damsky W, Galan A, McNiff JM. Revisiting histopathologic findings in Goltz syndrome. J Cutan Pathol. 2016;43:418–21. [PubMed: 26956940]
- Lasocki AL, Stark Z, Orchard D. A case of mosaic Goltz syndrome (focal dermal hypoplasia) in a male patient. Australas J Dermatol. 2011;52:48–51. [PubMed: 21332693]
- Liu J, Hsu PT, VanderWielen BA, Teng JM. Treatment of recalcitrant excessive granulation tissue with photodynamic therapy in an eight-year-old patient with focal dermal hypoplasia syndrome. Pediatr Dermatol. 2012;29:324–6. [PubMed: 21995324]
- Lombardi MP, Bulk S, Celli J, Lampe A, Gabbett MT, Ousager LB, van der Smagt JJ, Soller M, Stattin EL, Mannens MA, Smigiel R, Hennekam RC. Pathogenic variant update for the PORCN gene. Hum Mutat. 2011;32:723–8. [PubMed: 21472892]
- Lopez-Porras RF, Arroyo C, Soto-Vega E. Focal dermal hypoplasia with uterus bicornis and renal ectopia: case report and review of the literature. Case Rep Dermatol. 2011;3:158–63. [PMC free article: PMC3177835] [PubMed: 21941481]
- Maas SM, Lombardi MP, van Essen AJ, Wakeling EL, Castle B, Temple IK, Kumar VK, Writzl K, Hennekam RC. Phenotype and genotype in 17 patients with Goltz-Gorlin syndrome. J Med Genet. 2009;46:716–20. [PubMed: 19586929]
- Madan S, Liu W, Lu JT, Sutton VR, Toth B, Joe P, Waterson JR, Gibbs RA, Van den Veyver IB, Lammer EJ, Campeau PM, Lee BH. A non-mosaic PORCN mutation in a male with severe congenital anomalies overlapping focal dermal hypoplasia. Mol Genet Metab Rep. 2017;12:57–61. [PMC free article: PMC5466597] [PubMed: 28626639]
- Martinez-Campayo N, Rego-Campuzano I, del Pozo J, Paradela S, Fernandez-Flores A, Fonseca E. Novel uses of laser therapy in Goltz syndrome. Dermatol Ther. 2022;35:e15371. [PubMed: 35141996]
- Mary L, Scheidecker S, Kohler M, Lombardi MP, Delezoide AL, Auberger E, Triau S, Colin E, Gerard M, Grzeschik KH, Dollfus H, Antal MC. Prenatal diagnosis of focal dermal hypoplasia: Report of three fetuses and review of the literature. Am J Med Genet A. 2017 Feb;173(2):479–486. [PubMed: 27623003]
- Moog U, Jones MC, Bird LM, Dobyns WB. Oculocerebrocutaneous syndrome: the brain malformation defines a core phenotype. J Med Genet. 2005;42:913–21. [PMC free article: PMC1735958] [PubMed: 15879499]
- Motil KJ, Fete M, Fete TJ. Growth, nutritional, and gastrointestinal aspects of focal dermal hypoplasia (Goltz-Gorlin syndrome). Am J Med Genet C Semin Med Genet. 2016;172C:29–33. [PubMed: 27001925]
- Murakami C, de Oliveira Lira Ortega A, Guimaraes AS, Goncalves-Bittar D, Bonecker M, Ciamponi AL. Focal dermal hypoplasia: a case report and literature review. Oral surgery, oral medicine, oral pathology, oral radiology, and endodontics. 2011;112:e11–8. [PubMed: 21684779]
- Patrizi A, Tabanelli M, Grzeschik KH, Misciali C, Neri I, Happle R. Multiple basal cell carcinomas in a 38-year-old woman with Goltz syndrome. Dermatology. 2012;224:97–100. [PubMed: 22414489]
- Proffitt KD, Virshup DM. Precise regulation of porcupine activity is required for physiological Wnt signaling. J Biol Chem. 2012;287:34167–78. [PMC free article: PMC3464525] [PubMed: 22888000]
- Reddy J, Laufer MR. Congenital anomalies of the female reproductive tract in a patient with Goltz syndrome. J Pediatr Adolesc Gynecol. 2009;22:e71–2. [PubMed: 19646662]
- Rhee KY, Baek RM, Ahn KJ. Airway management in a patient with focal dermal hypoplasia. Anesth Analg. 2006;103:1342. [PubMed: 17056997]
- Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, Grody WW, Hegde M, Lyon E, Spector E, Voelkerding K, Rehm HL, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17:405–24. [PMC free article: PMC4544753] [PubMed: 25741868]
- Rosen SA, Bocklage T. Mucocutaneous squamous papilloma with reactive lymphoid hyperplasia in two patients with focal dermal hypoplasia. Pediatr Dev Pathol. 2005;8:250–2. [PubMed: 15747104]
- Ross J, Busch J, Mintz E, Ng D, Stanley A, Brafman D, Sutton VR, Van den Veyver I, Willert K. A rare human syndrome provides genetic evidence that WNT signaling is required for reprogramming of fibroblasts to induced pluripotent stem cells. Cell Rep. 2014;9:1770–80. [PMC free article: PMC4335800] [PubMed: 25464842]
- Satcher KG, Maegawa GHB, Schoch JJ. Microphthalmia and linear skin defects syndrome: Precise diagnosis guides prognosis. Pediatr Dermatol. 2020;37:217–18. [PubMed: 31373408]
- Selzer G, David R, Revach M, Cvibah TJ, Fried A. Goltz syndrome with multiple giant-cell tumor-like lesions in bones. Ann Intern Med. 1974;80:714–7. [PubMed: 4364933]
- Shimaoka Y, Hatamochi A, Hamasaki Y, Shimura N, Arisaka O, Imai Y, Yamazaki S. Severe focal dermal hypoplasia in a female patient transmitted from a mildly affected mother. J Dermatol. 2009;36:181–3. [PubMed: 19335698]
- Smith A, Hunt TR 3rd. The orthopedic characterization of Goltz syndrome. Am J Med Genet C Semin Med Genet. 2016;172C:41–3. [PubMed: 26867035]
- Tadini G, Brena M, Gelmetti C, Pezzani L. Goltz syndrome. In: Atlas of Genodermatoses. 2 ed. CRC Press; 2015:274-5.
- Takada R, Satomi Y, Kurata T, Ueno N, Norioka S, Kondoh H, Takao T, Takada S. Monounsaturated fatty acid modification of Wnt protein: its role in Wnt secretion. Dev Cell. 2006;11:791–801. [PubMed: 17141155]
- Tanaka H, Yasui N, Kuriskaki E, Shimomura Y. The Goltz syndrome associated with giant cell tumour of bone. A case report. Int Orthop. 1990;14:179–81. [PubMed: 2373565]
- Tanaka K, Okabayashi K, Asashima M, Perrimon N, Kadowaki T. The evolutionarily conserved porcupine gene family is involved in the processing of the Wnt family. Eur J Biochem. 2000;267:4300–11. [PubMed: 10866835]
- Tejani Z, Batra P, Mason C, Atherton D. Focal dermal hypoplasia: oral and dental findings. J Clin Pediatr Dent. 2005;30:67–72. [PubMed: 16302603]
- van Amerongen R, Nusse R. Towards an integrated view of Wnt signaling in development. Development. 2009;136:3205–14. [PubMed: 19736321]
- Vreeburg M, van Geel M, van den Heuij LG, Steijlen PM, van Steensel MA. Focal dermal hypoplasia in a male patient due to mosaicism for a novel PORCN single nucleotide deletion. J Eur Acad Dermatol Venereol. 2011;25:592–5. [PubMed: 20626533]
- Wang X, Sutton VR, Peraza-Llanes OJ, Yu Z, Rosetta R, Kou YC, Eble TN, Patel A, Thaller C, Fang P, Van den Veyver IB. Pathogenic variants in X-linked PORCN, a putative regulator of Wnt signaling, cause focal dermal hypoplasia. Nat Genet. 2007;39:836–8. [PubMed: 17546030]
- Wawrocka A, Walczak-Sztulpa J, Pawlak M, Gotz-Wieckowska A, Krawczynski MR. Non-syndromic anophthalmia/microphthalmia can be caused by a PORCN variant inherited in X-linked recessive manner. Am J Med Genet A. 2021;185:250–5. [PubMed: 33111437]
- Wechsler MA, Papa CM, Haberman F, Marion RW. Variable expression in focal dermal hypoplasia. An example of differential X-chromosome inactivation. Am J Dis Child. 1988;142:297–300. [PubMed: 3344717]
- Wright JT, Puranik CP, Farrington F. Oral phenotype and variation in focal dermal hypoplasia. Am J Med Genet C Semin Med Genet. 2016;172C:52–8. [PubMed: 26843121]
- Yesodharan D, Büschenfelde UMZ, Kutsche K, Mohandas Nair K, Nampoothiri S. Goltz-Gorlin syndrome: revisiting the clinical spectrum. Indian J Pediatr. 2018;85:1067–72. [PubMed: 29383603]
- Yoshihashi H, Ohki H, Torii C, Ishiko A, Kosaki K. Survival of a male mosaic for PORCN pathogenic variant with mild focal dermal hypoplasia phenotype. Pediatr Dermatol. 2011;28:550–4. [PubMed: 21133992]
Publication Details
Author Information and Affiliations
Baylor College of Medicine
Houston, Texas
Publication History
Initial Posting: May 15, 2008; Last Update: June 15, 2023.
Copyright
GeneReviews® chapters are owned by the University of Washington. Permission is hereby granted to reproduce, distribute, and translate copies of content materials for noncommercial research purposes only, provided that (i) credit for source (http://www.genereviews.org/) and copyright (© 1993-2024 University of Washington) are included with each copy; (ii) a link to the original material is provided whenever the material is published elsewhere on the Web; and (iii) reproducers, distributors, and/or translators comply with the GeneReviews® Copyright Notice and Usage Disclaimer. No further modifications are allowed. For clarity, excerpts of GeneReviews chapters for use in lab reports and clinic notes are a permitted use.
For more information, see the GeneReviews® Copyright Notice and Usage Disclaimer.
For questions regarding permissions or whether a specified use is allowed, contact: ude.wu@tssamda.
Publisher
University of Washington, Seattle, Seattle (WA)
NLM Citation
Sutton VR. PORCN-Related Developmental Disorders. 2008 May 15 [Updated 2023 Jun 15]. In: Adam MP, Feldman J, Mirzaa GM, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2024.



