Summary
Clinical characteristics.
Disorders of GNAS inactivation include the phenotypes pseudohypoparathyroidism Ia, Ib, and Ic (PHP-Ia, -Ib, -Ic), pseudopseudohypoparathyroidism (PPHP), progressive osseous heteroplasia (POH), and osteoma cutis (OC).
PHP-Ia and PHP-Ic are characterized by:
- End-organ resistance to endocrine hormones including parathyroid hormone (PTH), thyroid-stimulating hormone (TSH), gonadotropins (LH and FSH), growth hormone-releasing hormone (GHRH), and CNS neurotransmitters (leading to obesity and variable degrees of intellectual disability and developmental delay); and
- The Albright hereditary osteodystrophy (AHO) phenotype (short stature, round facies, and subcutaneous ossifications) and brachydactyly type E (shortening mainly of the 4th and/or 5th metacarpals and metatarsals and distal phalanx of the thumb).
Although PHP-Ib is characterized principally by PTH resistance, some individuals also have partial TSH resistance and mild features of AHO (e.g., brachydactyly).
PPHP, a more limited form of PHP-Ia, is characterized by various manifestations of the AHO phenotype without the hormone resistance or obesity.
POH and OC are even more restricted variants of PPHP:
- POH consists of dermal ossification beginning in infancy, followed by increasing and extensive bone formation in deep muscle and fascia.
- OC consists of extra-skeletal ossification that is limited to the dermis and subcutaneous tissues.
Diagnosis/testing.
The diagnosis of a disorder of GNAS inactivation is established in a proband with all or some of the characteristic clinical and endocrine findings and evidence on molecular genetic testing of a genetic or epigenetic alteration resulting in lack of expression/function of the GNAS complex locus.
PHP-Ia,.-Ib, and -Ic are associated with reduced or absent expression/function of the protein Gsα (encoded by the maternal GNAS complex locus) due to one of the following:
- An inactivating GNAS pathogenic variant
- A genetic alteration in the imprinting regulatory elements in the GNAS complex locus or the nearby gene, STX16, that prevents proper maternal imprint of the GNAS complex locus
- Isolated epimutations
- Paternal 20q disomy
PPHP and POH/OC phenotypes are associated with lack of expression/function of Gsα encoded by the paternal GNAS allele due to an inactivating GNAS pathogenic variant; the POH/OC phenotypes are also associated with lack of expression/function of Gsα (encoded by the maternal GNAS allele) as a result of an inactivating GNAS pathogenic variant.
Management.
Treatment of manifestations: Deficiencies of parathyroid hormone, thyroid hormone, and gonadotropins due to hormone resistance are treated in a standard manner. Growth hormone replacement therapy should be considered if screening for growth hormone deficiency with appropriate provocative testing is abnormal. Subcutaneous ossifications that are superficial and well circumscribed may be surgically removed when they are large or cause local irritation, although they may recur. Obesity tends to be the most difficult manifestation to treat as individuals with PHP-Ia and PHP-Ic have decreased resting energy expenditure and hyperphagia; thus, the usual recommendation of reduced caloric intake and increased physical activity may be less successful than in persons with obesity from other causes.
Surveillance: Routine monitoring of:
- Endocrine function: measurement of serum concentration of PTH, calcium and phosphate, TSH and free T4, and urinary calcium excretion;
- Growth velocity and growth hormone status (serum IGF1 and/or stimulated growth hormone testing);
- New and/or enlarging ectopic ossifications;
- Development of and/or progression of cataracts; and
- Psychoeducational needs regarding school assistance / educational support and developmental therapies (e.g., physical, occupational, and speech therapy).
Agents/circumstances to avoid: Limit dietary intake of phosphorus (dairy products and meats) in persons with persistently elevated serum levels of phosphate.
Evaluation of relatives at risk: It is appropriate to evaluate apparently asymptomatic first-degree relatives of an affected individual in order to identify as early as possible those who would benefit from prompt initiation of treatment.
Pregnancy management: For women with a disorder of GNAS inactivation that affects the maternal allele: Monitoring of serum concentration of calcium and thyroid studies (TSH, free T4) throughout pregnancy, labor, and the postpartum period and supplementation of calcium, vitamin D, and thyroid hormone as needed.
Genetic counseling.
Disorders of GNAS inactivation are inherited in an autosomal dominant manner with the specific phenotype determined by the parental origin of the defective allele. Of individuals with a disorder of GNAS inactivation, approximately 38% have an affected parent and 38% have a de novo GNAS pathogenic variant; in the remaining approximately 25% the cause is unknown.
Each child of an individual with a disorder of GNAS inactivation has a 50% chance of inheriting the parent's genetic alteration (except for simplex cases with PHP-1b for whom the mode of inheritance is not well established). If the maternal GNAS complex locus is affected, her offspring are at risk for PHP-Ia, PHP-Ib (when associated with deletions at the imprinting regulatory elements), or PHP-Ic; if the paternal allele has an inactivating GNAS pathogenic variant, his offspring are at risk for PPHP or POH/OC. If the genetic alteration in the GNAS complex locus or the GNAS pathogenic variant has been identified in an affected family member, prenatal testing for a pregnancy at increased risk and preimplantation genetic testing are technically possible.
GeneReview Scope
Table
Pseudohypoparathyroidism Ia (PHP-Ia) Pseudohypoparathyroidism Ib (PHP-Ib)
Diagnosis
No specific clinical criteria establish the diagnosis of a disorder of GNAS inactivation.
Suggestive Findings
A disorder of GNAS inactivation should be suspected in individuals with the following phenotypes.
Pseudohypoparathyroidism Ia (PHP-Ia) and pseudohypoparathyroidism Ic (PHP-Ic). The most readily recognized form of PHP is PHP-Ia, which has clinical and endocrine features similar to PHP-Ic. Note: PHP-Ic differs from PHP-Ia on the basis of normal functional activity of Gsα (the protein encoded by GNAS) determined in some biochemical assays based on receptor-independent activation of Gsα [Levine 2012].
PHP-Ia and PHP-Ic should be suspected in individuals with some of the following clinical and endocrine findings (which may emerge over time):
- End-organ resistance to several endocrine hormones:
- Parathyroid hormone (PTH), usually manifest as elevated PTH levels, hyperphosphatemia, and hypocalcemia, in the absence of vitamin D deficiency or magnesium deficiency
- Thyroid-stimulating hormone (TSH), manifest as hypothyroidism and elevated TSH levels in the absence of goiter or evidence of autoimmune thyroid disease
- Gonadotropins (LH and FSH), which may manifest in some females as reduced fertility and menstrual disorders/irregularities and in some males as cryptorchidism (often bilateral) and elevated LH and FSH levels.In some females, metabolic or endocrine disturbances may alter LH and FSH secretion to produce the biochemical appearance of hypothalamic amenorrhea.
- Growth hormone-releasing hormone (GHRH), manifest as growth hormone (hGH) deficiency with consequent poor growth and/or short stature, in 50% to 80% of the individuals tested. Note that IGF1 levels are often normal at diagnosis.
- Calcitonin, with asymptomatic hypercalcitonemia
- CNS neurotransmitters, leading to obesity and variable degrees of intellectual disability and developmental delayNote: Many affected individuals have normal neurocognitive function.
- Albright hereditary osteodystrophy (AHO) phenotype:
- Short stature
- Round facies
- Subcutaneous ossifications
- Brachydactyly type E (shortening mainly of the 4th and/or 5th metacarpals and metatarsals and distal phalanx of the thumb)Note: Shortened metacarpals may be recognized by the replacement of knuckles by dimples when making a fist.
- Intrauterine growth restriction
Pseudohypoparathyroidism Ib (PHP-Ib). Suggestive findings include:
- PTH resistance, the principal endocrine abnormality
- In some affected individuals:
- Partial TSH resistance with slightly elevated TSH levels and generally normal (or low) serum concentrations of thyroid hormones [Levine 2012]
- Mild brachydactyly (despite absence of the classic AHO phenotype)
- Enhanced intrauterine growth [Bréhin et al 2015] (See Table 2.)
- Madelung deformity [Sanchez et al 2011]
Pseudopseudohypoparathyroidism (PPHP), a more limited form of PHP-Ia, involves various manifestations of the AHO phenotype without hormone resistance or obesity.
Progressive osseous heteroplasia (POH), a more restricted variant of PPHP, consists of dermal ossification beginning in infancy, followed by increasing and extensive bone formation in deep muscle and fascia [Kaplan et al 1994].
Osteoma cutis (OC), another more restricted variant of PPHP, consists of extraskeletal ossification limited to the dermis and subcutaneous tissues.
Establishing the Diagnosis
The diagnosis of a disorder of GNAS inactivation is established in a proband with all or some of the above clinical findings and evidence on molecular genetic testing of a genetic or epigenetic alteration resulting in lack of expression/function of the GNAS complex locus. See Table 1 for a summary of molecular genetic testing, Table 2 for a summary of the phenotypes and genetic mechanisms of disorders of GNAS inactivation, and Molecular Genetics for details of the GNAS molecular defects.
The PHP-Ia, -Ib, and -Ic phenotypes are associated with lack of expression/function of the protein Gsα (encoded by the maternal GNAS complex locus) as a result of one of the following:
- An inactivating GNAS pathogenic variant
- A genetic alteration in the imprinting regulatory elements in the GNAS complex locus or the nearby gene, STX16, that prevents proper maternal imprint of the GNAS complex locus
- Isolated epimutations [Takatani et al 2015]
- Uniparental paternal 20q disomy [Takatani et al 2015]
The PPHP and POH/OC phenotypes are associated with lack of expression/function of Gsα (encoded by the paternal GNAS allele) due to an inactivating GNAS pathogenic variant; the POH/OC phenotypes are also associated with lack of expression/function of Gsα (encoded by the maternal GNAS allele) caused by an inactivating GNAS pathogenic variant.
Molecular genetic testing approaches can include a combination of gene-targeted testing (multigene panel, single-gene testing) and genomic testing (comprehensive genomic sequencing) depending on the phenotype.
Gene-targeted testing requires the clinician to determine which gene(s) are likely involved, whereas genomic testing may not. Because of the varied manifestations of the disorders of GNAS inactivation, individuals with the findings of one of the distinctive phenotypes described in Suggestive Findings are likely to be diagnosed using gene-targeted testing (see Option 1), whereas those with clinical findings indistinguishable from other inherited disorders with similar endocrine abnormalities are more likely to be diagnosed using genomic testing (see Option 2).
Option 1
When the clinical and endocrine findings suggest one of the distinctive phenotypes, molecular genetic testing approaches can include single-gene testing and methylation analysis or use of a multigene panel.
Single-gene testing and methylation analysis
- Single-gene testing. Sequence analysis of exons 1 through 13 of GNAS is performed first, followed by gene-targeted deletion/duplication analysis if no pathogenic variant is found.Note: In persons with the PHP-Ib phenotype, STX16 deletion/duplication analysis should be performed if no GNAS pathogenic variant is identified and if methylation defects are limited to GNAS exon A/B differentially methylated region (DMR) (also referred to as exon 1A or GNAS A/B:TSS-DMR).
- Methylation analysis examines differentially methylated regions (DMRs) of the GNAS complex locus for loss of the normal methylation pattern on the maternal allele (i.e., an imprinting defect). When a GNAS pathogenic variant is not identified on sequence analysis, it is appropriate to perform methylation analysis. Note: Although loss of methylation can identify the presence of an imprinting defect, it cannot identify the cause of the imprinting defect.PHP-Ia. In some instances, individuals with PHP-Ia may have partial methylation defects of uncertain significance.PHP-Ib. Because alterations of various imprinting control regions are associated with PHP-Ib, loss of methylation at the exon A/B DMR of the maternal GNAS complex locus is observed in all affected individuals; thus, methylation analysis should be the initial test in individuals with findings suggestive of PHP-1b.
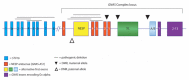
- Familial PHP-Ib is in most instances caused either by multiexon deletions disrupting the upstream gene STX16 or (less frequently) by deletions involving NESP (Figure 1) [Elli et al 2014a].
- Sporadic PHP-Ib. The genetic basis for the methylation defect in sporadic PHP-Ib is usually unknown; however, broad GNAS imprinting abnormalities involving multiple DMRs have been observed in most affected individuals, some of whom had molecular genetic findings consistent with paternal uniparental 20q isodisomy [Takatani et al 2015] (Table 2).Note: in some cases, deletions encompassing the whole GNAS complex locus can mimic methylation defects associated with sporadic PHP-Ib. In case of an overall methylation defect, deletion/duplication analysis should be performed.
Multigene panel
- A multigene panel that includes GNAS and other genes of interest (see Differential Diagnosis) may also be considered. Note: (1) The genes included in the panel and the diagnostic sensitivity of the testing used for each gene vary by laboratory and are likely to change over time. (2) Some multigene panels may include genes not associated with the condition discussed in this GeneReview; thus, clinicians need to determine which multigene panel is most likely to identify the genetic cause of the condition while limiting identification of variants of uncertain significance and pathogenic variants in genes that do not explain the underlying phenotype. (3) In some laboratories, panel options may include a custom laboratory-designed panel and/or custom phenotype-focused exome analysis that includes genes specified by the clinician. (4) Methods used in a panel may include sequence analysis, deletion/duplication analysis, and/or other non-sequencing-based tests. These approaches do not allow the characterization of imprinting defects.
Option 2
When the phenotype is indistinguishable from other inherited disorders with similar endocrine abnormalities, comprehensive genomic testing (exome sequencing or genome sequencing) can be considered.
For an introduction to comprehensive genomic testing click here. More detailed information for clinicians ordering genomic testing can be found here.
Note: Genomic testing does not allow characterization of imprinting defects.
Table 1.
Molecular Genetic Testing Used in Disorders of GNAS Inactivation
Clinical Characteristics
Clinical Description
Disorders of GNAS inactivation include pseudohypoparathyroidism Ia (PHP-Ia), Ib (PHP-Ib), and Ic (PHP-Ic), as well as pseudopseudohypoparathyroidism (PPHP), progressive osseous heteroplasia (POH), and osteoma cutis (OC) (see Table 2).
The term pseudohypoparathyroidism (PHP) refers to disorders with hypocalcemia and hyperphosphatemia (which are typical of hypoparathyroidism) that result from end-organ resistance to ‒ rather than deficiency of ‒ parathyroid hormone (PTH).
Pseudohypoparathyroidism Ia (PHP-Ia) and PHP-Ic
PHP-Ia and PHP-Ic have a similar phenotype and are distinguished only by ex vivo assays of Gsα protein function that are based on hormone receptor activation of Gsα; in these assays Gsα activity is reduced by approximately 50% in PHP-Ia and normal in PHP-Ic (Table 2).
Endocrine. The clinical phenotype consists of metabolic resistance to multiple endocrine hormones including parathyroid hormone (PTH), thyroid-stimulating hormone (TSH), growth hormone-releasing hormone (GHRH), calcitonin, and often gonadotropins [Levine 2012]. The most common endocrinopathies are biochemical hypoparathyroidism, primary hypothyroidism (without goiter), and growth hormone deficiency (demonstrated in 50%-70% of affected individuals) [de Sanctis et al 2007]. Obesity, due to decreased resting energy expenditure [Roizen et al 2016], appears to result from impaired Gsα-coupled signaling in imprinted regions of the hypothalamus (which, based on mouse studies, is likely to be in the dorsomedial hypothalamus) [Chen et al 2017].
The development of the endocrine features occurs over time. The earliest manifestation of hormone resistance is usually mild hypothyroidism, which is often discovered during newborn screening as an elevated TSH with normal serum levels of thyroid hormones. The average age at diagnosis of PHP-Ia is around age seven years, when PTH resistance and hypocalcemia are recognized; however, individuals with milder manifestations may not be diagnosed until the third decade of life [Linglart et al 2013, Turan & Bastepe 2015].
- TSH resistance is variable and may manifest as congenital hypothyroidism; however, TSH may be elevated in individuals who are euthyroid, or only after formal TRH stimulation testing. Serum levels of thyroid hormone are usually normal or only mildly depressed.
- PTH resistance. Although random PTH levels are often elevated in PHP-Ia, hypocalcemia may fluctuate and may not become clinically significant until later childhood. Hyperphosphatemia usually precedes hypocalcemia. Some individuals remain eucalcemic.Occasionally, early PTH resistance is associated with hypercalcemia rather than hypocalcemia. Presumably, in these individuals the kidney retains the ability to increase production of 1,25(OH)2D, the active form of vitamin D, in response to the elevated levels of circulating PTH that result from decreased ability to excrete urinary phosphate [MA Levine, personal observation].As in PTH-deficient hypoparathyroidism, protracted hypocalcemia and hyperphosphatemia lead to ectopic calcification, particularly in the brain at the grey-white cerebral intersection and the basal ganglia and ocular lenses, manifesting as posterior subcapsular cataracts.Bone density is often increased [Long et al 2010].Unrecognized hypocalcemia may present with tetany, seizures, or laryngeal spasm; seizures appear to be more common, independent of hypocalcemia [Fitch 1982]. Cataracts can also be seen in the setting of prolonged hypocalcemia.
- Gonadotropin resistance may result in delayed puberty and incomplete development of secondary sex characteristics, menstrual irregularities, or reduced fertility.
- Early-onset morbid obesity can begin in infancy [Dekelbab et al 2009]. At least 65% of those younger than age 18 years are obese [Fitch 1982].
Sleep apnea is also common (45% of individuals), but is only partly explained by obesity, as individuals with PHP-Ia have a fourfold increased risk for sleep apnea compared to similarly obese individuals. This increase may be due to hypotonia as well as the effects of Gsα inactivation on the normal sleep cycle [Landreth et al 2015].
Musculoskeletal. At birth growth parameters are usually normal, but can be below normal [Wilson 2006]; however, neonates may then present with congenital hypothyroidism or ectopic ossifications.
Linear growth may initially be normal or advanced due to obesity; bone age is frequently advanced beyond chronologic age [Fitch 1982]. Linear growth slows soon after early childhood and prematurely ceases in early puberty, leading to height below the third percentile in the majority of adults [Fitch 1982, Wilson 2006] primarily due to premature closure of epiphyseal growth plates; however, GH deficiency may also be a contributing factor.
Affected individuals display clinical features of Albright hereditary osteodystrophy (AHO) comprising a round face, short stature, brachydactyly/brachymetacarpia, and heterotopic ossification of the dermis and subcutaneous tissues. Macrocephaly relative to height is typical: 40% have a head circumference above the 90th percentile [Fitch 1982, Wilson 2006].
The most common musculoskeletal feature is brachydactyly; brachydactyly type E is manifest as shortened metacarpals (particularly 4th and 5th) and metatarsals (particularly 3rd and 4th) plus brachydactyly type D, manifest as shortening of the distal phalanx of the thumb.
Even in the absence of brachymetacarpia, individuals with AHO usually have brachydactyly type D, a shortened distal thumb phalanx and short, broad thumbnails, associated radiographically with cone-shaped epiphyses [Fitch 1982].
Brachydactyly may lead to carpal tunnel syndrome with symptomatic paresthesia, which may be confused with the symptoms of hypocalcemia [Joseph et al 2011].
Other reported musculoskeletal features include craniosynostosis, hyperostosis of the cranial vault, absence of normal caudal widening of the lumbar interpedicular distances (associated with spinal stenosis), ossification of paravertebral ligaments, shortened distal ulnas, bowing of the tibia and radius, small capital femoral epiphyses, coxa vara, coxa valga, increased prevalence of bony exostoses, and carpal tunnel syndrome [Wilson & Hall 2002, van Lindert et al 2008, Joseph et al 2011].
Skin. Ectopic ossifications (also known as osteoma cutis) are true heterotopic intramembraneous bone which occur in 60%-70% of affected individuals, independent of calcium, phosphate, or PTH levels [Prendiville et al 1992].
Ectopic ossifications are most commonly cutaneous, either within subdermal fat or in the dermis. They are located most commonly in the scalp and extremities (particularly the periarticular areas of the hands and feet). These lesions may be very small and asymptomatic, or painful; occasionally lesions can extrude a chalky material. Removal of ectopic ossifications does not result in progression or exacerbation although they may recur if removal is incomplete [Fitch 1982, Prendiville et al 1992, Wilson 2006].
Other areas of ectopic ossification include the sclera and choroid of the eye and cardiac ventricular septum. Visceral involvement is rare [Fitch 1982].
Dental changes such as enamel hypoplasia, widened root canals, shortened roots with open apices, thickened laminar dura, and delayed dental eruption have been noted in more than 30% of affected individuals, often with impacted second molars [Ritchie 1965].
Mild-to-moderate intellectual disability is seen in up to 79% of affected individuals [Mouallem et al 2008].
Pseudohypoparathyroidism Ib (PHP-Ib)
PHP-Ib consists of PTH resistance with partial resistance to TSH in some affected individuals. Partial TSH resistance manifests as slightly elevated serum TSH levels with generally normal (or low) serum concentrations of thyroid hormones [Levine 2012].
Poorly treated PTH resistance can lead to hyperparathyroid bone disease or tertiary hyperparathyroidism. Very rarely bone density can be elevated [Sbrocchi et al 2011].
Patterns of excessive growth or weight gain have been described in newborns or during early infancy and childhood.
Individuals with PHP-Ib may have growth-plate defects such as mild brachydactyly or a Madelung deformity-like defect [Sanchez et al 2011], but lack the complete constellation of features seen in AHO. Intellect is typically normal.
The average age of diagnosis for symptomatic individuals is age ten to 12 years [Linglart et al 2007], which is later than for PHP-Ia.
Pseudopseudohypoparathyroidism (PPHP)
The phenotype is heterogeneous with a wide differential diagnosis (see Differential Diagnosis).
Intrauterine growth restriction is common.
Ectopic ossifications are frequent and almost pathognomonic of Gsα deficiency.
Individuals with PPHP have normocalcemia and no endocrine defects, but have the physical phenotype of Albright hereditary osteodystrophy.
Obesity and intellectual disabilities (10%) are less prevalent than in PHP-Ia [Long et al 2007, Mouallem et al 2008].
Progressive Osseous Heteroplasia (POH)
Individuals with POH have no endocrine defects or features of AHO, but have progressive ectopic ossification that extends to deep connective tissues, often with debilitating effects [Levine 2012, Pignolo et al 2015].
Of note, POH-like ossifications have been observed rarely in individuals with AHO or PHP-1a.
Osteoma Cutis (OC)
Individuals with OC develop ossification limited to the dermis and subcutaneous tissues.
Phenotypes and Genetic Mechanisms of Disorders of GNAS Inactivation
Table 2 summarizes the different phenotypes that result from disorders of GNAS inactivation. Note the various molecular defects and associated parental origin. See Molecular Genetics and Figure 1 for a description and map of the GNAS complex locus and the upstream gene, STX16.
Table 2.
Phenotypes and Genetic Mechanisms of Disorders of GNAS Inactivation
Genotype-Phenotype Correlations
No clear correlation appears to exist between the type and location of GNAS complex locus / STX16 pathogenic variants and disease onset, severity of endocrine resistance, or number of AHO features.
However, two unique variants affecting both the stability and the activity of Gsα have been described in three unrelated individuals with PHP1a who presented with additional clinical features reflecting enhanced Gsα activity:
- A missense variant associated with typical PHP1a features as well as testotoxicosis [Nakamoto et al 1996]
- A four amino-acid insertion within the Gsα GDP/GTP-binding site identified in a brother and sister with PHP1a with transient neonatal diarrhea and pancreatic insufficiency who inherited the insertion from their unaffected mother, who had germline mosaicism [Aldred et al 2000]. Biochemical and intact cell studies suggested that the phenotype results from Gsα deficiency due to instability of the mutated protein and that the accompanying neonatal diarrhea may result from its enhanced constitutive activity in the intestine [Makita et al 2007].
Penetrance
Disorders of GNAS inactivation show complete penetrance, with manifestations typically appearing during childhood. However, the exact manifestations and severity vary significantly among individuals.
Prevalence
The estimated prevalence for pseudohypoparathyroidism (PHP) and Albright hereditary osteodystrophy (AHO) is approximately 0.79 per 100,000 (according to the Orphanet Report Series, November 2008).
A Japanese study estimated the prevalence of PHP at 3.4 per 1 million individuals [Nakamura et al 2000].
The prevalence of POH has never been estimated. It is likely extremely rare: fewer than 60 clinically confirmed individuals worldwide have been reported [Shore & Kaplan 2010].
Genetically Related (Allelic) Disorders
Postzygotic GNAS somatic pathogenic variants that result in a gain of function in Gsα can be seen in:
- McCune-Albright syndrome, which is characterized by the clinical triad of fibrous dysplasia (monostotic or polyostotic), café au lait macules with irregular borders (so-called "coast of Maine"), and autonomous endocrine function, commonly precocious puberty but also often including other endocrine abnormalities such as thyrotoxicosis, pituitary gigantism, and Cushing syndrome [Lumbroso et al 2004]. McCune-Albright syndrome is clinically heterogeneous due to the extent of the various tissues affected in the mosaic state;
- Isolated features of McCune-Albright syndrome (e.g., monostotic or polyostotic fibrous dysplasia), ovarian cysts and testicular sex cord tumors, acromegaly due to sporadic growth hormone-secreting adenomas in the pituitary, and ACTH-independent macronodular adrenal hyperplasia (none of which show other features of McCune-Albright syndrome); as well as intraductal papillary mucinous neoplasms of the pancreas [Furukawa et al 2011].
Note that heterozygous germline activating GNAS pathogenic variants are presumed to be lethal to the embryo.
Differential Diagnosis
Conditions to be considered in the differential diagnosis include the following:
- 2q37 deletion syndrome, which is characterized by mild-moderate developmental delay/intellectual disability, brachydactyly of digits 3-5 (often digit 4 alone), short stature, obesity, hypotonia, round facies, behavioral abnormalities, joint hypermobility/dislocation, and scoliosis. Affected individuals have normal endocrine function, however. In most individuals with the 2q37 deletion syndrome, the deletion is de novo. This condition can also be associated with heterozygous pathogenic variants in HDAC4.
- Idiopathic or primary hypoparathyroidism, resulting from decreased parathyroid function, can have many causes including hypo- or aplasia of the parathyroid gland or autoimmune-related damage. Symptoms include cramping and twitching of the muscles (tetany), paresthesias, fatigue, and abdominal pain. PTH and calcium levels are low and phosphorous levels are elevated.
- Kenny-Caffey syndrome (OMIM 244460) is characterized by proportionate short stature, cortical thickening and medullary stenosis of the tubular bones, delayed closure of the anterior fontanelle, transient hypocalcemia (due to primary hypoparathyroidism), and normal intelligence. An autosomal dominant form is caused by mutation of FAM111A (see FAM111A-Related Skeletal Dysplasias) and an autosomal recessive form by mutation of TBCE (formerly KCS1).
- Type E brachydactyly (OMIM 113300, 613382) shows wide variability with shortening mainly of the metacarpals and metatarsals, but in some cases the phalanges as well. Some individuals have moderate short stature and round facies but do not have ectopic ossification or intellectual disability. Heterozygous pathogenic variants in HOXD13 and PTHLH have been found in several affected individuals.
- Acrodysostosis (OMIM 101800, 614613) is characterized by short stature, early-onset diffuse brachydactyly, round face, nasal hypoplasia, advanced bone age, and obesity. Laboratory studies may show resistance to multiple hormones, including PTH (parathyroid hormone), TSH (thyroid-stimulating hormone), calcitonin, growth hormone-releasing hormone (GHRH), and gonadotropins (LH and FSH). Heterozygous pathogenic variants in PRKAR1A or PDE4D have been identified in some affected individuals.
Management
Evaluations Following Initial Diagnosis
To establish the extent of disease and needs in an individual diagnosed with a disorder of GNAS inactivation, the following evaluations are recommended:
- Assessment of height, weight, body mass index, growth velocity, and pubertal development
- X-ray of hands and feet to classify the type and extent of brachydactyly; bone age study to determine whether skeletal maturation is advanced
- Endocrinology evaluation, with studies that may include the following:
- Serum concentration of calcium and phosphorous
- Serum concentration of 25-hydroxyvitamin D and magnesium to evaluate respectively for severe vitamin D deficiency and hypomagnesemia, either of which can cause decreased responsiveness to PTH or secondary hyperparathyroidism
- Parathyroid hormone (PTH)
- Thyroid-stimulating hormone (TSH) and free T4
- Growth hormone evaluation (IGF1, IGFBP3, stimulated GH testing)
- Urinary calcium excretion and tubular reabsorption of phosphorusNote: Renal responsiveness to PTH (via measurement of the serum or urinary nephrogenous cyclic AMP response to administered PTH) is rarely indicated.
- Psychoeducational profile/developmental assessment
- Ophthalmologic examination for cataracts
- Consultation with nutritionist when obesity is present
- Consultation with a clinical geneticist and/or genetic counselor
Treatment of Manifestations
Hypocalcemia. Therapy of hypocalcemia is similar to the treatment of other forms of hypoparathyroidism: restoring serum calcium to the normal level using supplemental calcium (1-2 grams or 25-50 mg/kg of elemental calcium per day) and activated forms of vitamin D (calcitriol or 1-alpha calcidiol).
For all forms of PHP-I, calcium and activated vitamin D doses should be adjusted to try to maintain PTH levels at the upper limit of normal or even slightly higher (e.g., 50-150 pg/mL) to avoid secondary complications of chronic hyperparathyroidism. Urinary calcium excretion should be monitored to detect hypercalciuria; however, because calcium reabsorption in the thick ascending limb of the kidney remains responsive to PTH, hypercalciuria in these individuals is very rare as long as PTH is not suppressed.
Rarely thiazide diuretics can be considered in individuals who develop hypercalciuria.
Maintenance of a normal serum calcium-phosphorus product is recommended to minimize the risk of cataract formation and intracerebral calcification. Oral calcium supplements should be taken with meals to reduce serum phosphorus levels. Occasionally, phosphate-binding resins may be required.
Note: Because individuals with PHP-I do not respond to PTH, there is no role for PTH therapy in these individuals.
Hypothyroidism and gonadotropin resistance are treated in the conventional manner.
Growth hormone deficiency. If screening for growth hormone deficiency with appropriate provocative testing is abnormal, growth hormone replacement therapy should be considered. Note that in small case series treatment with growth hormone replacement increased growth velocity, but did not improve final adult height [Mantovani et al 2010]. Nevertheless, there are suggestions that growth hormone replacement may also have a salutary effect on weight. Failure to improve final adult height may be due to partial resistance to the paracrine effects of PTH-related peptides in growth plate cartilage, accounting for advanced skeletal maturation, premature closure of the epiphyses, and short stature [Bastepe et al 2004].
Subcutaneous ossifications may be surgically removed if they are large or cause local irritation. Ossifications may recur if residua are left in situ. Ossifications may enlarge in individuals treated with growth hormone.
Obesity is the greatest treatment challenge. Current strategies involve the usual recommendations of reduced caloric intake and increased physical activity. Given that individuals with PHP-Ia and PHP-Ic have decreased resting energy expenditure and hyperphagia, these efforts may be less successful than in persons with obesity from other causes.
Surveillance
Surveillance includes the following:
- Once the diagnosis is established, annual monitoring for endocrine abnormalities with measurement of serum concentration of PTH, calcium, and phosphate; TSH and free T4, and urinary calcium excretion (either 24-hour urine collection or random urine collections for determination of the calcium/creatinine ratio). These studies should be begun as soon an individual begins treatment, and may be performed more frequently in growing children who may experience increasing requirements for thyroid hormone and/or activated forms of vitamin D.
- Growth velocity and growth hormone status (serum IGF1 and/or stimulated growth hormone testing) should be evaluated annually. Individuals who receive growth hormone replacement should be monitored every three to four months per customary protocols.
- Routine physical examination including assessment of: (1) height to identify changes in growth velocity; (2) the hands and feet for evidence of brachydactyly; and (3) new and/or enlarging ectopic ossifications
- Annual examination by an ophthalmologist to monitor development of and/or progression of cataracts
- Periodic assessment of psychoeducational needs regarding school assistance/educational support and developmental therapies (e.g., physical, occupational, and speech therapy)
- Monitoring of post-pubertal females for disturbances in hypothalamic-pituitary-ovarian function
Agents/Circumstances to Avoid
Limit dietary intake of phosphorus (dairy products and meats) in persons with persistently elevated serum levels of phosphate.
Evaluation of Relatives at Risk
It is appropriate to evaluate apparently asymptomatic first-degree relatives of an affected individual in order to identify as early as possible those who would benefit from initiation of treatment. Evaluations can include:
- Molecular genetic testing if the genetic mechanism of GNAS inactivation has been identified in the family (Table 2).
- If the genetic mechanism of GNAS inactivation is not known, at-risk relatives can be assessed by physical examination and regular measurement of serum concentrations of parathyroid hormone (PTH), calcium, and phosphorus.
See Genetic Counseling for issues related to testing of at-risk relatives for genetic counseling purposes.
Pregnancy Management
Serum concentration of calcium and thyroid studies (TSH and free T4) should be monitored throughout pregnancy, labor, and the postpartum period.
Calcium, vitamin D, and thyroid hormone should be supplemented as needed [Singh et al 2012]. Note that requirements for thyroid hormone will increase in early pregnancy, while requirements for activated forms of vitamin D may decline during the third trimester.
Because breastfeeding can significantly reduce requirements for activated vitamin D, mothers should be monitored for this during lactation and after weaning.
Therapies Under Investigation
Dr Emily Germain-Lee at the University of Connecticut is recruiting individuals with PHP-Ia and AHO to determine whether growth hormone therapy can improve short stature and obesity.
Researchers at Vanderbilt University Medical Center are investigating whether theophylline treatment promotes weight loss, improves glucose tolerance, and slows growth plate closure in children and young adults.
Search ClinicalTrials.gov in the US and EU Clinical Trials Register in Europe for access to information on clinical studies for a wide range of diseases and conditions.
Genetic Counseling
Genetic counseling is the process of providing individuals and families with information on the nature, mode(s) of inheritance, and implications of genetic disorders to help them make informed medical and personal decisions. The following section deals with genetic risk assessment and the use of family history and genetic testing to clarify genetic status for family members; it is not meant to address all personal, cultural, or ethical issues that may arise or to substitute for consultation with a genetics professional. —ED.
Mode of Inheritance
Disorders of GNAS inactivation are inherited in an autosomal dominant manner with the specific phenotype determined by the parental origin of the defective allele [Lemos &Thakker 2015].
Pseudohypoparathyroidism (PHP) Ia, Ib, and Ic result from reduced or absent expression/function of the protein Gsα (encoded by the maternal GNAS complex locus) due to one of the following:
- An inactivating GNAS pathogenic variant
- A genetic alteration in the imprinting regulatory elements in the GNAS complex locus or the nearby gene, STX16, that prevents proper maternal imprint of the GNAS complex locus
- Isolated epimutations
- Paternal 20q disomy
The PPHP and POH/OC phenotypes are associated with lack of expression/function of Gsα encoded by the paternal GNAS allele due to an inactivating GNAS pathogenic variant; the POH/OC phenotypes are associated with lack of expression/function of Gsα encoded by the maternal GNAS allele due to an inactivating GNAS pathogenic variant.
Risk to Family Members
Of individuals with a disorder of GNAS inactivation, approximately 38% have an affected parent and 38% have a de novo GNAS pathogenic variant; in the remaining approximately 25% the cause is unknown [Elli et al 2016].
Table 3.
Disorders of GNAS Inactivation: Risk to Sibs of a Proband Based on the Parents' Genetic Status
Offspring of a proband with a disorder of GNAS inactivation. Each child of an individual with a disorder of GNAS inactivation has a 50% chance of inheriting the genetic alteration, except for persons with simplex PHP-1b, in which there may not be an underlying heritable variant. The phenotype of the offspring who inherit a GNAS pathogenic variant or imprinting defect depends on the sex of the parent of origin (see Table 4).
Table 4.
Disorder of GNAS Inactivation: Phenotypes for Offspring Based on the Sex of the Parent
Note regarding inheritance of PHP-Ib: Although females who inherit a PHP-Ib-related genetic alteration from their father (i.e., a paternally imprinted PHP-Ib-related genetic alteration) will be unaffected, their offspring have a 50% risk of inheriting the now maternally imprinted PHP-Ib-related genetic alteration and being affected.
Other family members. The risk to other family members depends on the status of the proband's parents: if a parent is affected, the parent's family members may be at risk.
Related Genetic Counseling Issues
See Management, Evaluation of Relatives at Risk for information on evaluating at-risk relatives for the purpose of early diagnosis and treatment.
Considerations in families with an apparent de novo pathogenic variant. When neither parent of a proband with an autosomal dominant condition has the pathogenic variant identified in the proband or clinical evidence of the disorder, the pathogenic variant is likely de novo. However, non-medical explanations including alternate paternity or maternity (e.g., with assisted reproduction) and undisclosed adoption could also be explored.
Family planning
- The optimal time for determination of genetic risk and discussion of the availability of prenatal/preimplantation genetic testing is before pregnancy.
- It is appropriate to offer genetic counseling (including discussion of potential risks to offspring and reproductive options) to young adults who are affected or at risk.
DNA banking. Because it is likely that testing methodology and our understanding of genes, pathogenic mechanisms, and diseases will improve in the future, consideration should be given to banking DNA from probands in whom a molecular diagnosis has not been confirmed (i.e., the causative pathogenic mechanism is unknown). For more information, see Huang et al [2022].
Prenatal Testing and Preimplantation Genetic Testing
Once the mechanism of GNAS inactivation has been identified in an affected family member, prenatal testing for a pregnancy at increased risk for a disorder of GNAS inactivation and preimplantation genetic testing are possible.
The phenotype in offspring who inherit a GNAS pathogenic variant depends on the sex of the parent of origin (i.e., the parent from whom an individual inherited the GNAS pathogenic variant; see Risk to Family Members, Offspring of a proband with a disorder of GNAS inactivation).
Differences in perspective may exist among medical professionals and within families regarding the use of prenatal testing. While most centers would consider use of prenatal testing to be a personal decision, discussion of these issues may be helpful.
Resources
GeneReviews staff has selected the following disease-specific and/or umbrella support organizations and/or registries for the benefit of individuals with this disorder and their families. GeneReviews is not responsible for the information provided by other organizations. For information on selection criteria, click here.
- Metabolic Support UKUnited KingdomPhone: 0845 241 2173
- UCLA International Skeletal Dysplasia Registry (ISDR)Phone: 310-825-8998
Molecular Genetics
Information in the Molecular Genetics and OMIM tables may differ from that elsewhere in the GeneReview: tables may contain more recent information. —ED.
Table A.
Disorders of GNAS Inactivation: Genes and Databases
Table B.
OMIM Entries for Disorders of GNAS Inactivation (View All in OMIM)
Molecular Pathogenesis
The PHP-Ia, Ib, Ic phenotypes are associated with lack of expression/function of the protein Gsα (encoded by the maternal GNAS complex locus) as a result of one of the following:
- An inactivating GNAS pathogenic variant
- A genetic alteration in the imprinting regulatory elements in the GNAS complex locus or the nearby gene, STX16, that prevents proper maternal imprint of the GNAS complex locus
- Isolated epimutations
- Paternal 20q disomy
PPHP is associated with lack of expression/function of Gsα encoded from the paternal GNAS allele with an inactivating GNAS pathogenic variant. POH/OC is also most commonly associated with paternal GNAS pathogenic variants.
Gene Structure
GNAS is a complex transcriptional unit with multiple transcript variants through the use of alternative first exons, alternative splicing of downstream exons, antisense transcripts, and reciprocal imprinting. For detailed summaries of gene, transcript, and protein isoform information see: Figure 1; Table A, Gene; Levine [2012]; Pignolo et al [2015]; Mantovani et al [2016]; and Tafaj & Jüppner [2017].
Gsα is encoded by GNAS exons 1-13 from the transcript variant NM_000516.5, which is expressed from both maternal and paternal alleles in most cells. However, in some cells (e.g., pituitary somatotropes, proximal renal tubular cells, thyroid epithelial cells, and gonadal cells) Gsα is primarily expressed from the maternal allele; preferential maternal expression may also occur in other tissues. While the Gsα promoter is not methylated, it appears that cis-acting elements that control tissue-specific paternal imprinting of Gsα are located within the primary imprint region in exon A/B (also referred to as exon 1A) [Levine 2012] (see Figure 1).
Pathogenic Variants
PHP-Ia and PHP-Ic and PPHP or POH/OC. Although the inactivating GNAS pathogenic variants observed in the phenotypes PHP-Ia and PHP-Ic and the phenotypes PPHP or POH/OC are similar, they differ in the parent of origin (Table 2):
- PHP-Ia and PHP-Ic result from lack of expression of the maternal allele
- PPHP result from lack of expression of the paternal allele.
- POH and OC can be associated with pathogenic variants in either the maternal or paternal allele; however, paternal pathogenic variants are more common.
PHP-Ib. The genetic alteration differs in familial cases and simplex cases (i.e., a single occurrence in a family) (Table 2).
Familial PHP-Ib genetic alterations include:
- A pathogenic GNAS variant in exon 13 on the maternal allele;
- Loss of imprinting (methylation) at the maternal GNAS exon A/B DMR (also referred to as exon 1A or GNAS A/B:TSS-DMR);
- Deletion of maternal STX16 exons 3-5 or 4-6 [Bastepe et al 2003, Linglart et al 2005];
- Deletion of maternal NESP and/or NESP-AS [Bastepe et al 2005, Chillambhi et al 2010, Richard et al 2012, Rezwan et al 2015];
- Deletion of maternal STX16 [Elli et al 2014b, Tafaj & Jüppner 2017].
Simplex PHP-Ib genetic alterations include:
- GNAS imprinting abnormalities that involve multiple DMRs [Mantovani et al 2012];
- Paternal uniparental disomy for all or part of chromosome 20 [Fernandez-Rebollo et al 2010, Linglart et al 2013].
General comments. The most commonly found GNAS variants are frameshift, nonsense, or splicing; they are expected to lead to untranslated proteins. Nonsense and single-nucleotide variants that alter translation initiation or mRNA splicing have also been documented [Elli et al 2013a, Elli et al 2016].
Heterozygous GNAS pathogenic variants in exon 1 are the most common (18%). Only one individual has been described with a pathogenic variant in exon 3, which is alternatively spliced out of transcripts that encode a slightly smaller form of Gsα [Thiele et al 2007]. While pathogenic variants are distributed across GNAS [Mantovani et al 2016], exon 7 is a hot spot for the 4-bp GNAS deletion c.565_568delGACT [Elli et al 2013a].
Individuals with pathogenic variants in exon 1 of GNAS have a higher prevalence of ectopic ossifications than other affected individuals [Elli et al 2013a]. Truncating variants are associated with more ectopic ossifications than other types of pathogenic variants [Lemos & Thakker 2015, Thiele et al 2015].
Table 5.
GNAS Pathogenic Variants Discussed in This GeneReview
Normal Gene Product
GNAS encodes the alpha subunit of the stimulatory guanine nucleotide-binding protein (Gsα), which binds and hydrolyzes GTP as a downstream effect following activation of a hormone receptor [Elli et al 2013b].
GNAS encodes for two protein isoforms depending on whether exon 3 is included (52-kd) or excluded (45-kd).
Abnormal Gene Product
Haploinsufficiency of Gsα results in PHP-Ia and PHP-Ic (lack of expression of the maternal GNAS allele) and PPHP (lack of expression of the paternal GNAS allele).
In PHP-Ib, the imprinting defects lead to reduced expression of Gsα in a tissue-specific manner.
In PHP-Ic heterozygous GNAS pathogenic variants do not affect the function of Gsα (in vitro activity is normal), but rather affect the coupling of Gsα to its receptors. In a few affected individuals, pathogenic variants in exon 13 were shown to affect receptor-mediated activation [Mantovani et al 2012]. The discussion of PHP-1c can be complicated: patients with Gs coupling defects in fact have PHP-1a and are misassigned due to the vagaries of the old ex vivo assay, which did not activate Gs via a receptor pathway. PHP-1c should be reserved for the rare persons (if they exist) who have PHP-1a and no observable Gs abnormality.
Chapter Notes
Author Notes
Dr Levine's web page
Revision History
- 26 October 2017 (bp) Review posted live
- 15 April 2015 (mal) Original submission
References
Literature Cited
- Ahrens W, Hiort O, Staedt P, Kirschner T, Marschke C, Kruse K. Analysis of the GNAS1 gene in Albright's hereditary osteodystrophy. J Clin Endocrinol Metab. 2001;86:4630–4. [PubMed: 11600516]
- Aldred MA, Aftimos S, Hall C, Waters KS, Thakker RV, Trembath RC, Brueton L. Constitutional deletion of chromosome 20q in two patients affected with albright hereditary osteodystrophy. Am J Med Genet. 2002;113:167–72. [PubMed: 12407707]
- Aldred MA, Bagshaw RJ, Macdermot K, Casson D, Murch SH, Walker-Smith JA, Trembath RC. Germline mosaicism for a GNAS1 mutation and Albright hereditary osteodystrophy. J Med Genet. 2000;37:E35. [PMC free article: PMC1734481] [PubMed: 11073544]
- Bastepe M, Fröhlich LF, Hendy GN, Indridason OS, Josse RG, Koshiyama H, Körkkö J, Nakamoto JM, Rosenbloom AL, Slyper AH, Sugimoto T, Tsatsoulis A, Crawford JD, Jüppner H. Autosomal dominant pseudohypoparathyroidism type Ib is associated with a heterozygous microdeletion that likely disrupts a putative imprinting control element of GNAS. J Clin Invest. 2003;112:1255–63. [PMC free article: PMC213493] [PubMed: 14561710]
- Bastepe M, Fröhlich LF, Linglart A, Abu-Zahra HS, Tojo K, Ward LM, Jüppner H. Deletion of the NESP55 differentially methylated region causes loss of maternal GNAS imprints and pseudohypoparathyroidism type Ib. Nat Genet. 2005;37:25–7. [PubMed: 15592469]
- Bastepe M, Weinstein LS, Ogata N, Kawaguchi H, Jüppner H, Kronenberg HM, Chung UI. Stimulatory G protein directly regulates hypertrophic differentiation of growth plate cartilage in vivo. Proc Natl Acad Sci U S A. 2004;101:14794–9. [PMC free article: PMC522030] [PubMed: 15459318]
- Bréhin AC, Colson C, Maupetit-Méhouas S, Grybek V, Richard N, Linglart A, Kottler ML, Jüppner H. Loss of methylation at GNAS exon A/B is associated with increased intrauterine growth. J Clin Endocrinol Metab. 2015;100:E623–31. [PMC free article: PMC4399294] [PubMed: 25603460]
- Chen M, Shrestha YB, Podyma B, Cui Z, Naglieri B, Sun H, Ho T, Wilson EA, Li YQ, Gavrilova O, Weinstein LS. Gsα deficiency in the dorsomedial hypothalamus underlies obesity associated with Gsα mutations. J Clin Invest. 2017;127:500–10. [PMC free article: PMC5272175] [PubMed: 27991864]
- Chillambhi S, Turan S, Hwang DY, Chen HC, Jüppner H, Bastepe M. Deletion of the noncoding GNAS antisense transcript causes pseudohypoparathyroidism type Ib and biparental defects of GNAS methylation in cis. J Clin Endocrinol Metab. 2010;95:3993–4002. [PMC free article: PMC2913043] [PubMed: 20444925]
- Dekelbab BH, Aughton DJ, Levine MA. Pseudohypoparathyroidism type 1a and morbid obesity in infancy. Endocr Pract. 2009;15:249–53. [PubMed: 19364695]
- de Sanctis L, Bellone J, Salerno M, Faleschini E, Caruso-Nicoletti M, Cicchetti M, Concolino D, Balsamo A, Buzi F, Ghizzoni L, de Sanctis C. GH secretion in a cohort of children with pseudohypoparathyroidism type Ia. J Endocrinol Invest. 2007;30:97–103. [PubMed: 17392598]
- Elli FM, Barbieri AM, Bordogna P, Ferrari P, Bufo R, Ferrante E, Giardino E, Beck-Peccoz P, Spada A, Mantovani G. Screening for GNAS genetic and epigenetic alterations in progressive osseous heteroplasia: first Italian series. Bone. 2013a;56:276–80. [PubMed: 23796510]
- Elli FM, de Sanctis L, Bollati V, Tarantini L, Filopanti M, Barbieri AM, Peverelli E, Beck-Peccoz P, Spada A, Mantovani G. Quantitative analysis of methylation defects and correlation with clinical characteristics in patients with pseudohypoparathyroidism type I and GNAS epigenetic alterations. J Clin Endocrinol Metab. 2014a;99:E508–17. [PubMed: 24423294]
- Elli FM, deSanctis L, Ceoloni B, Barbieri AM, Bordogna P, Beck-Peccoz P, Spada A, Mantovani G. Pseudohypoparathyroidism type Ia and pseudo-pseudohypoparathyroidism: the growing spectrum of GNAS inactivating mutations. Hum Mutat. 2013b;34:411–6. [PubMed: 23281139]
- Elli FM, de Sanctis L, Peverelli E, Bordogna P, Pivetta B, Miolo G, Beck-Peccoz P, Spada A, Mantovani G. Autosomal dominant pseudohypoparathyroidism type Ib: a novel inherited deletion ablating STX16 causes loss of imprinting at the A/B DMR. J Clin Endocrinol Metab. 2014b;99:E724–8. [PubMed: 24438374]
- Elli FM, Linglart A, Garin I, de Sanctis L, Bordogna P, Grybek V, Pereda A, Giachero F, Verrua E, Hanna P, Mantovani G, Perez de Nanclares G. The prevalence of GNAS deficiency-related diseases in a large cohort of patients characterized by the EuroPHP Network. J Clin Endocrinol Metab. 2016;101:3657–68. [PubMed: 27428667]
- Fernandez-Rebollo E, García-Cuartero B, Garin I, Largo C, Martínez F, Garcia-Lacalle C, Castaño L, Bastepe M, Pérez de Nanclares G. Intragenic GNAS deletion involving exon A/B in pseudohypoparathyroidism type 1A resulting in an apparent loss of exon A/B methylation: potential for misdiagnosis of pseudohypoparathyroidism type 1B. J Clin Endocrinol Metab. 2010;95:765–71. [PMC free article: PMC2840867] [PubMed: 20008020]
- Fitch N. Albright's hereditary osteodystrophy: a review. Am J Med Genet. 1982;11:11–29. [PubMed: 6278930]
- Furukawa T, Kuboki Y, Tanji E, Yoshida S, Hatori T, Yamamoto M, Shibata N, Shimizu K, Kamatani N, Shiratori K. Whole-exome sequencing uncovers frequent GNAS mutations in intraductal papillary mucinous neoplasms of the pancreas. Sci Rep. 2011;1:161. [PMC free article: PMC3240977] [PubMed: 22355676]
- Garin I, Elli FM, Linglart A, Silve C, de Sanctis L, Bordogna P, Pereda A, Clarke JT, Kannengiesser C, Coutant R, Tenebaum-Rakover Y, Admoni O, de Nanclares GP, Mantovani G. Novel microdeletions affecting the GNAS locus in pseudohypoparathyroidism: characterization of the underlying mechanisms. J Clin Endocrinol Metab. 2015;100:E681–7. [PubMed: 25594858]
- Geneviève D, Sanlaville D, Faivre L, Kottler ML, Jambou M, Gosset P, Boustani-Samara D, Pinto G, Ozilou C, Abeguilé G, Munnich A, Romana S, Raoul O, Cormier-Daire V, Vekemans M. Paternal deletion of the GNAS imprinted locus (including Gnasxl) in two girls presenting with severe pre- and post-natal growth retardation and intractable feeding difficulties. Eur J Hum Genet. 2005;13:1033–9. [PubMed: 15915160]
- Huang SJ, Amendola LM, Sternen DL. Variation among DNA banking consent forms: points for clinicians to bank on. J Community Genet. 2022;13:389–97. [PMC free article: PMC9314484] [PubMed: 35834113]
- Joseph AW, Shoemaker AH, Germain-Lee EL. Increased prevalence of carpal tunnel syndrome in Albright hereditary osteodystrophy. J Clin Endocrinol Metab. 2011;96:2065–73. [PMC free article: PMC3135204] [PubMed: 21525160]
- Kaplan FS, Craver R, MacEwen GD, Gannon FH, Finkel G, Hahn G, Tabas J, Gardner RJ, Zasloff MA. Progressive osseous heteroplasia: a distinct developmental disorder of heterotopic ossification. Two new case reports and follow-up of three previously reported cases. J Bone Joint Surg Am. 1994;76:425–36. [PubMed: 8126048]
- Landreth H, Malow BA, Shoemaker AH. Increased prevalence of sleep apnea in children with pseudohypoparathyroidism Type 1a. Horm Res Paediatr. 2015;84:1–5. [PMC free article: PMC4540680] [PubMed: 25925491]
- Lemos MC, Thakker RV. GNAS mutations in pseudohypoparathyroidism type 1a and related disorders. Hum Mutat. 2015;36:11–9. [PMC free article: PMC4309471] [PubMed: 25219572]
- Levine MA. An update on the clinical and molecular characteristics of pseudohypoparathyroidism. Curr Opin Endocrinol Diabetes Obes. 2012;19:443–51. [PMC free article: PMC3679535] [PubMed: 23076042]
- Linglart A, Bastepe M, Juppner H. Similar clinical and laboratory findings inpatients with symptomatic autosomal dominant and sporadic autosomal dominant and sporadic pseudohypoparathyroidism type 1b despite different epigenetic changes at the GNAS locus. Clin Endocrinol (Oxf). 2007;67:822–31. [PubMed: 17651445]
- Linglart A, Carel JC, Garabédian M, Lé T, Mallet E, Kottler ML. GNAS1 lesions in pseudohypoparathyroidism Ia and Ic: genotype phenotype relationship and evidence of the maternal transmission of the hormonal resistance. J Clin Endocrinol Metab. 2002;87:189–97. [PubMed: 11788646]
- Linglart A, Gensure RC, Olney RC, Jüppner H, Bastepe M. A novel STX16 deletion in autosomal dominant pseudohypoparathyroidism type Ib redefines the boundaries of a cis-acting imprinting control element of GNAS. Am J Hum Genet. 2005;76:804–14. [PMC free article: PMC1199370] [PubMed: 15800843]
- Linglart A, Maupetit-Mehouas S, Silve C. GNAS-related loss-of-function disorders and the role of imprinting. Horm Res Paediatr. 2013;79:119–29. [PubMed: 23548772]
- Long DN, Levine MA, Germain-Lee EL. Bone mineral density in pseudohypoparathyroidism type 1a. J Clin Endocrinol Metab. 2010;95:4465–75. [PMC free article: PMC2936057] [PubMed: 20610593]
- Long DN, McGuire S, Levine MA, Weinstein LS, Germain-Lee EL. Body mass index differences in pseudohypoparathyroidism type 1a versus pseudopseudohypoparathyroidism may implicate paternal imprinting of Galpha(s) in the development of human obesity. J Clin Endocrinol Metab. 2007;92:1073–9. [PubMed: 17164301]
- Lumbroso S, Paris F, Sultan C., European Collaborative Study. Activating Gsalpha mutations: analysis of 113 patients with signs of McCune-Albright syndrome – a European Collaborative Study. J Clin Endocrinol Metab. 2004;89:2107–13. [PubMed: 15126527]
- Makita N, Sato J, Rondard P, Fukamachi H, Yuasa Y, Aldred MA, Hashimoto M, Fujita T, Iiri T. Human G(salpha) mutant causes pseudohypoparathyroidism type Ia/neonatal diarrhea, a potential cell-specific role of the palmitoylation cycle. Proc Natl Acad Sci U S A. 2007;104:17424–9. [PMC free article: PMC2077272] [PubMed: 17962410]
- Mantovani G, Elli FM, Spada A. GNAS epigenetic defects and pseudohypoparathyroidism: time for a new classification? Horm Metab Res. 2012;44:716–23. [PubMed: 22674477]
- Mantovani G, Ferrante E, Giavoli C, Linglart A, Cappa M, Cisternino M, Maghnie M, Ghizzoni L, de Sanctis L, Lania AG, Beck-Peccoz P, Spada A. Recombinant human GH replacement therapy in children with pseudohypoparathyroidism type 1a: first study on the effect on growth. J Clin Endocrinol Metab. 2010;95:5011–7. [PubMed: 20719837]
- Mantovani G, Spada A, Elli FM. Pseudohypoparathyroidism and Gsα–cAMP-linked disorders: current view and open issues. Nat Rev Endocrinol. 2016;12:347–56. [PubMed: 27109785]
- Mitsui T, Naaki K, Takagi M, Narumi S, Ishii T, Hasegawa T. A family of pseudohypoparathyroidism type Ia with an 850-kb submicroscopic deletion encompassing the whole GNAS locus. Am J Med Genet A. 2012;158A:261–4. [PubMed: 22140064]
- Mouallem M, Shaharabany M, Weintrob N, Shalitin S, Nagelberg N, Shapira H, Zadik Z, Farfel Z. Cognitive impairment is prevalent in pseudohypoparathyroidism type 1a, but not in pseudopseudohypoparathyroidism: possible cerebral imprinting of Gsalpha. Clin Endocrinol (Oxf). 2008;68:233–9. [PubMed: 17803690]
- Nakamoto JM, Zimmerman D, Jones EA, Loke KY, Siddiq K, Donlan MA, Brickman AS, Van Dop C. Concurrent hormone resistance (pseudohypoparathyroidism type Ia) and hormone independence (testotoxicosis) caused by a unique mutation in the G alpha s gene. Biochem Mol Med. 1996;58:18–24. [PubMed: 8809352]
- Nakamura Y, Matsumoto T, Tamakoshi A, Kawamura T, Seino Y, Kasuga M, Yanagawa H, Ohno Y. Prevalence of idiopathic hypoparathyroidism and pseudohypoparathyroidism in Japan. J Epidemiol. 2000;10:29–33. [PubMed: 10695258]
- Ngai YF, Chijiwa C, Mercimek-Mahmutoglu S, Stewart L, Yong SL, Robinson WP, Gibson WT. Pseudohypoparathyroidism type 1a and the GNAS p.R231H mutation: Somatic mosaicism in a mother with two affected sons. Am J Med Genet A. 2010;152A:2784–90. [PubMed: 20979189]
- Pignolo RJ, Ramaswamy G, Fong JT, Shore EM, Kaplan FS. Progressive osseous heteroplasia: diagnosis, treatment, and prognosis. Appl Clin Genet. 2015;8:37–48. [PMC free article: PMC4321643] [PubMed: 25674011]
- Prendiville JS, Lucky AW, Mallory SB, Mughal Z, Mimouni F, Langman CB. Osteoma cutis as a presenting sign of pseudohypoparathyroidism. Pediatr Dermatol. 1992;9:11–8. [PubMed: 1574470]
- Rezwan FI, Poole RL, Prescott T, Walker JM, Karen Temple I, Mackay DJ. Very small deletions within the NESP55 gene in pseudohypoparathyroidism type 1b. Eur J Hum Genet. 2015;23:494–9. [PMC free article: PMC4247793] [PubMed: 25005734]
- Richard N, Abeguilé G, Coudray N, Mittre H, Gruchy N, Andrieux J, Cathebras P, Kottler ML. A new deletion ablating NESP55 causes loss of maternal imprint of A/B GNAS and autosomal dominant pseudohypoparathyroidism type Ib. J Clin Endocrinol Metab. 2012;97:E863–7. [PubMed: 22378814]
- Richard N, Molin A, Coudray N, Rault-Guillaume P, Jüppner H, Kottler ML. Paternal GNAS mutations lead to severe intrauterine growth retardation (IUGR) and provide evidence for a role of XLαs in fetal development. J Clin Endocrinol Metab. 2013;98:E1549–56. [PMC free article: PMC3763972] [PubMed: 23884777]
- Ritchie GM. Dental manifestations of pseudohypoparathyroidism. Arch Dis Child. 1965;40:565–72. [PMC free article: PMC2019454] [PubMed: 5830003]
- Roizen JD, Danzig J, Groleau V, McCormack S, Casella A, Harrington J, Sochett E, Tershakovec A, Zemel BS, Stallings VA, Levine MA. Resting energy expenditure is decreased in pseudohypoparathyroidism type 1A. J Clin Endocrinol Metab. 2016;2016;101:880–8. [PMC free article: PMC4803160] [PubMed: 26709970]
- Sanchez J, Perera E, Jan de Beur S, Ding C, Dang A, Berkovitz GD, Levine MA. Madelung-like deformity in pseudohypoparathyroidism type 1b. J Clin Endocrinol Metab. 2011;96:E1507–11. [PMC free article: PMC3167675] [PubMed: 21752878]
- Sbrocchi AM, Rauch F, Lawson ML, Hadjiyannakis S, Lawrence S, Bastepe M, Jüppner H, Ward LM. Osteosclerosis in two brothers with autosomal dominant pseudohypoparathyroidism type 1b: bone histomorphometric analysis. Eur J Endocrinol. 2011;2011;164:295–301. [PMC free article: PMC3810006] [PubMed: 21062889]
- Shore EM, Ahn J, Jan de Beur S, Li M, Xu M, Gardner RJ, Zasloff MA, Whyte MP, Levine MA, Kaplan FS. Paternally inherited inactivating mutations of the GNAS1 gene in progressive osseous heteroplasia. N Engl J Med. 2002;346:99–106. [PubMed: 11784876]
- Shore EM, Kaplan FS. Inherited human diseases of heterotopic bone formation. Nat Rev Rheumatol. 2010;6:518–27. [PMC free article: PMC3551620] [PubMed: 20703219]
- Singh A, Agarwal N, Chopra S, Sikka P, Suri V, Kumar B, Dutta P. Management of pseudohypoparathyroidism type 1a during pregnancy and labor: a case report. Case Rep Obstet Gynecol. 2012;2012:629583. [PMC free article: PMC3388577] [PubMed: 22779017]
- Tafaj O, Jüppner H. Pseudohypoparathyroidism: one gene, several syndromes. J Endocrinol Invest. 2017;40:347–56. [PubMed: 27995443]
- Takatani R, Minagawa M, Molinaro A, Reyes M, Kinoshita K, Takatani T, Kazukawa I, Nagatsuma M, Kashimada K, Sato K, Matsushita K, Nomura F, Shimojo N, Jüppner H. Similar frequency of paternal uniparental disomy involving chromosome 20q (patUPD20q) in Japanese and Caucasian patients affected by sporadic pseudohypoparathyroidism type Ib (sporPHP1B). Bone. 2015;79:15–20. [PMC free article: PMC4501871] [PubMed: 25997889]
- Thiele S, Werner R, Ahrens W, Hoppe U, Marschke C, Staedt P, Hiort O. A disruptive mutation in exon 3 of the GNAS gene with Albright hereditary osteodystrophy, normocalcemic pseudohypoparathyroidism, and selective long transcript variant Gsalpha-l deficiency. J Clin Endocrinol Metab. 2007;92:1764–8. [PubMed: 17299070]
- Thiele S, Werner R, Grötzinger J, Brix B, Staedt P, Struve D, Reiz B, Farida J, Hiort O. A positive genotype-phenotype correlation in a large cohort of patients with pseudohypoparathyroidism type ia and pseudo-pseudohypoparathyroidism and 33 newly identified mutations in the GNAS gene. Mol Genet Genomic Med. 2015;3:111–20. [PMC free article: PMC4367083] [PubMed: 25802881]
- Turan S, Bastepe M. GNAS spectrum of disorders. Curr Osteoporos Rep. 2015;13:146–58. [PMC free article: PMC4417430] [PubMed: 25851935]
- van Lindert EJ, Bartels RH, Noordam K. Spinal stenosis with paraparesis in albright hereditary osteodystrophy. Case report and review of the literature. Pediatr Neurosurg. 2008;44:337–40. [PubMed: 18552518]
- Wilson LC. Albright's hereditary osteodystrophy. J Pediatr Endocrinol Metab. 2006;19 Suppl 2:671–3. [PubMed: 16789633]
- Wilson LC, Hall CM. Albright's hereditary osteodystrophy and pseudohypoparathyroidism. Semin Musculoskelet Radiol. 2002;6:273–83. [PubMed: 12541184]
Publication Details
Author Information and Affiliations
Asheville, North Carolina
Children's Hospital of Philadelphia;
University of Pennsylvania Perelman School of Medicine
Philadelphia, Pennsylvania
Publication History
Initial Posting: October 26, 2017.
Copyright
GeneReviews® chapters are owned by the University of Washington. Permission is hereby granted to reproduce, distribute, and translate copies of content materials for noncommercial research purposes only, provided that (i) credit for source (http://www.genereviews.org/) and copyright (© 1993-2024 University of Washington) are included with each copy; (ii) a link to the original material is provided whenever the material is published elsewhere on the Web; and (iii) reproducers, distributors, and/or translators comply with the GeneReviews® Copyright Notice and Usage Disclaimer. No further modifications are allowed. For clarity, excerpts of GeneReviews chapters for use in lab reports and clinic notes are a permitted use.
For more information, see the GeneReviews® Copyright Notice and Usage Disclaimer.
For questions regarding permissions or whether a specified use is allowed, contact: ude.wu@tssamda.
Publisher
University of Washington, Seattle, Seattle (WA)
NLM Citation
Haldeman-Englert CR, Hurst ACE, Levine MA. Disorders of GNAS Inactivation. 2017 Oct 26. In: Adam MP, Feldman J, Mirzaa GM, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2024.
