Summary
Clinical characteristics.
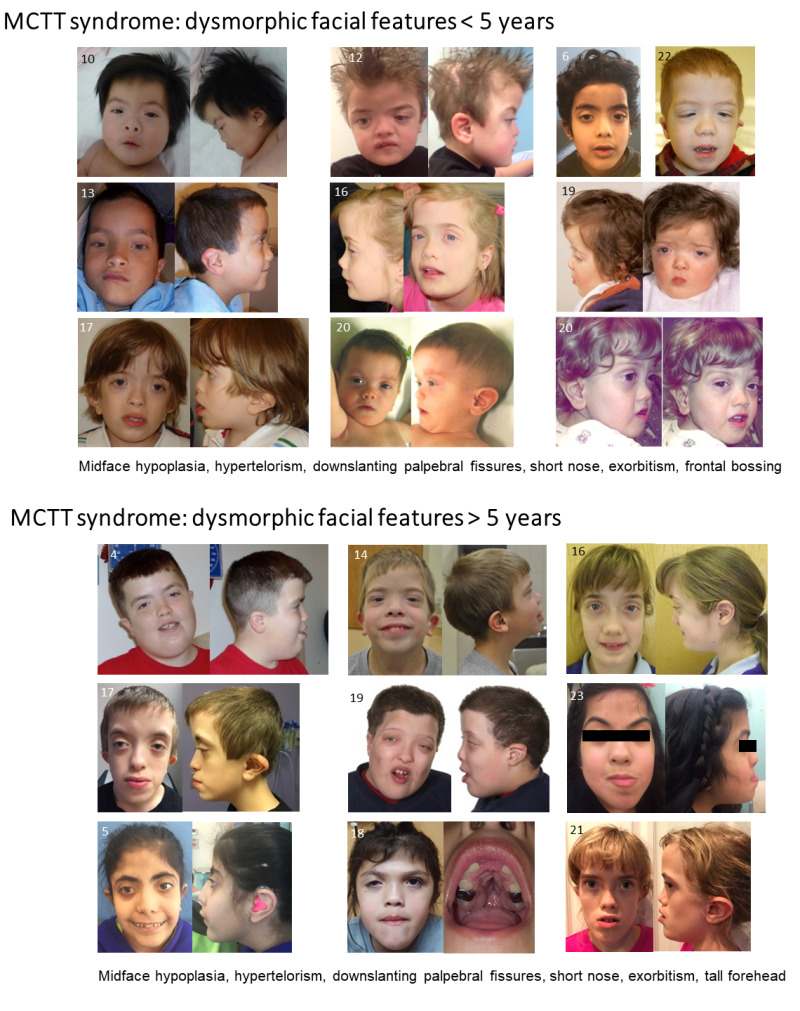
Individuals with MN1 C-terminal truncation (MCTT) syndrome have mild-to-moderate intellectual disability, severe expressive language delay, dysmorphic facial features (midface hypoplasia, downslanting palpebral fissures, hypertelorism, exophthalmia, short upturned nose, and small low-set ears), and distinctive findings on brain imaging (including perisylvian polymicrogyria and atypical rhombencephalosynapsis). Mild-to-moderate prelingual hearing loss (usually bilateral, conductive, and/or sensorineural) is common. Generalized seizures (observed in the minority of individuals) are responsive to anti-seizure medication. There is an increased risk for craniosynostosis and, thus, increased intracranial pressure. To date, 25 individuals with MCTT syndrome have been identified.
Diagnosis/testing.
No consensus clinical diagnostic criteria for MCTT syndrome have been published. The diagnosis is established in a proband with suggestive findings and a heterozygous pathogenic variant in MN1 identified by molecular genetic testing.
Management.
Treatment of manifestations: Multidisciplinary specialists to help manage developmental delay / intellectual disability, feeding issues, seizures, hearing loss, and speech and language needs, especially alternative communication.
Surveillance: Routine follow up by multidisciplinary specialists per their recommendations.
Genetic counseling.
MCTT syndrome is an autosomal dominant disorder typically caused by a de novo MN1 pathogenic variant. The risk to the sibs of a proband depends on the genetic status of the proband's parents: if the MN1 pathogenic variant found in the proband cannot be detected in the leukocyte DNA of either parent, the recurrence risk to sibs is estimated to be slightly greater than that of the general population because of the possibility of parental somatic/germline mosaicism. Prenatal testing for a pregnancy at increased risk and preimplantation genetic testing are possible once the MN1 pathogenic variant has been identified in an affected family member.
Diagnosis
No consensus clinical diagnostic criteria for MN1 C-terminal truncation (MCTT) syndrome have been published.
Suggestive Findings
MN1 C-terminal truncation (MCTT) syndrome should be suspected in individuals with the following clinical findings and brain MRI findings.
Clinical findings
- Intellectual disability (ID) with severe expressive language delay
- Hypotonia
- Delays in motor development
- Hearing loss (conductive or sensorineural)
- Distinctive craniofacial features (See Figure 1.)

Brain MRI findings. In MCTT syndrome, an atypical distinctive form of rhombencephalosynapsis is observed in which there is partial or complete loss of the cerebellar vermis landmarks with fusion of the cerebellar hemispheres characterized by abnormal midline crossing of cerebellar folia and white matter tracts. Other distinctive brain MRI findings are summarized in Table 1 (see Figure 2 and Figure 3).
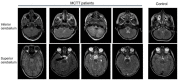
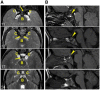
Table 1.
Brain Imaging Features in Individuals with MCTT Syndrome
Establishing the Diagnosis
The diagnosis of MN1 C-terminal truncation (MCTT) syndrome is established in a proband with suggestive findings and a heterozygous pathogenic (or likely pathogenic) variant in MN1 identified by molecular genetic testing (see Table 2).
(1) Per ACMG/AMP variant interpretation guidelines, the terms "pathogenic variants" and "likely pathogenic variants" are synonymous in a clinical setting, meaning that both are considered diagnostic and both can be used for clinical decision making [Richards et al 2015]. Reference to "pathogenic variants" in this section is understood to include any likely pathogenic variants. (2) Identification of a heterozygous MN1 variant of uncertain significance does not establish or rule out the diagnosis of this disorder.
Molecular genetic testing approaches can include a combination of gene-targeted testing (single-gene testing and multigene panel) and comprehensive genomic testing (exome sequencing, genome sequencing) depending on the phenotype.
Gene-targeted testing requires that the clinician determine which gene(s) are likely involved, whereas genomic testing does not. Individuals with the distinctive craniofacial or brain MRI findings described in Suggestive Findings are likely to be diagnosed using gene-targeted testing (see Option 1), whereas those in whom the diagnosis of MCTT syndrome has not been considered are more likely to be diagnosed using genomic testing (see Option 2).
Option 1
Single-gene testing. Sequence analysis of MN1 is performed first to detect small intragenic deletions/insertions and missense, nonsense, and splice site variants. Note: Depending on the sequencing method used, single-exon, multiexon, or whole-gene deletions/duplications may not be detected. Typically, if no variant is detected by the sequencing method used, the next step is to perform gene-targeted deletion/duplication analysis to detect exon and whole-gene deletions or duplications; however, since MCTT occurs through a gain-of-function mechanism and large intragenic deletion or duplication has not been reported, testing for intragenic deletions or duplication is much less likely to identify a disease-causing variant given the current understanding.
Note: To date all described variants have been C-terminal truncating variants at the 3' end of exon 1 or in exon 2.
An intellectual disability multigene panel that includes MN1 and other genes of interest (see Differential Diagnosis) is most likely to identify the genetic cause of the condition while limiting identification of variants of uncertain significance and pathogenic variants in genes that do not explain the underlying phenotype. Note: (1) The genes included in the panel and the diagnostic sensitivity of the testing used for each gene vary by laboratory and are likely to change over time. (2) Some multigene panels may include genes not associated with the condition discussed in this GeneReview. Given that MCTT syndrome is quite rare and only recently identified, some intellectual disability panels may not include MN1. (3) In some laboratories, panel options may include a custom laboratory-designed panel and/or custom phenotype-focused exome analysis that includes genes specified by the clinician. (4) Methods used in a panel may include sequence analysis, deletion/duplication analysis, and/or other non-sequencing-based tests.
For an introduction to multigene panels click here. More detailed information for clinicians ordering genetic tests can be found here.
Option 2
Comprehensive genomic testing does not require the clinician to determine which gene(s) are likely involved. Exome sequencing is most commonly used; genome sequencing is also possible.
For an introduction to comprehensive genomic testing click here. More detailed information for clinicians ordering genomic testing can be found here.
Table 2.
Molecular Genetic Testing Used in MN1 C-Terminal Truncation Syndrome
Clinical Characteristics
Clinical Description
To date, 25 individuals have been identified with a germline (de novo or inherited from a mosaic parent) pathogenic C-terminal variant in MN1 [Mak et al 2020, Miyake et al 2020]. Individuals with MN1 C-terminal truncation (MCTT) syndrome have intellectual disability, severe expressive language delay, dysmorphic facial features (see Figure 1), and distinctive findings on brain imaging (including perisylvian polymicrogyria and atypical rhombencephalosynapsis) (see Table 1). The summary of the phenotypic features associated with MCTT syndrome in Table 3 is based on these reports.
Table 3.
Clinical Features of MN1 C-Terminal Truncation Syndrome
Developmental delay (DD) / intellectual disability (ID). Individuals with MCTT syndrome have mild-to-moderate intellectual disability and severe expressive language delay. The majority have nonverbal communication with notable exceptions. For example, one child was able to speak at age two years, and at age 14 years was able to function at the level of a seven-year-old. For others, single-word speech began between ages three and six years. Some can communicate in sign language with up to 50 signs [Mak et al 2020].
Delay in gross motor development included hypotonia; at least four of 22 children walked independently by age two to three years. Others required orthotics or a wheelchair for mobilization. As for fine motor and self-help skills, most individuals require help with writing, feeding, or dressing [Mak et al 2020].
Hearing loss, when present, is mild to moderate and prelingual. It is usually bilateral, conductive, and/or sensorineural. Dysplasia of the cochlea, semicircular canals, and bony structures of the middle ear (e.g., incus) has been reported.
Feeding difficulties are more prominent early in infancy and may resolve after the first year of life. Hyperphagia has also been reported in three children [Miyake et al 2020].
Spinal anomalies include lordosis, scoliosis, or kyphosis, which may be detected clinically or by imaging.
Dental anomalies can include conical teeth, crowded teeth, and serrated teeth. Malocclusion has also been reported.
Ophthalmologic anomalies can include oculomotor defects, strabismus, and/or shallow orbits, giving the appearance of exorbitism.
Cardiovascular anomalies include atrial septal defect or ventricular septal defects.
Seizures are generalized and may be myoclonic in nature. Among the six individuals with seizures, the majority were isolated events and were responsive to anti-seizure medication. While polymicrogyria may be associated with increased risk of seizures, more information is needed.
Craniosynostosis. Individuals with MCTT syndrome are at increased risk for craniosynostosis with no specific pattern to the sutures affected. Although head circumference is normal in individuals with craniosynostosis, head shape is consistently affected. Skull shape anomalies that may be observed with or without underlying craniosynostosis include dolichocephaly, turricephaly, and/or bitemporal narrowing, plagiocephaly, and macrocephaly. Individuals with craniosynostosis are at increased risk for elevated intracranial pressure.
Behavioral problems. While some affected individuals may experience frustration due to poor verbal communication, no consistent behavior problems have been reported. The natural history into adulthood has not yet been delineated.
Growth. Growth parameters tend to remain within the normal range.
Prognosis. It is unknown if life span is affected in MCTT syndrome. One individual is alive at age 21 years [Mak et al 2020]. An unreported male is well at age 39 years [Angela Lin, personal commmunication], demonstrating that survival into adulthood is possible. Since many adults with disabilities have not undergone advanced genetic testing, it is likely that adults with this condition are unrecognized and underreported.
Genotype-Phenotype Correlations
No genotype-phenotype correlations have been identified.
Nomenclature
MCTT syndrome is referred to as CEBALID (craniofacial defects, dysmorphic ears, structural brain abnormalities, expressive language delay, and impaired intellectual development) syndrome in OMIM (OMIM 618774).
Prevalence
To date, 25 individuals with MCTT syndrome have been reported [Mak et al 2020, Miyake et al 2020]. This disorder is expected to be rare; the prevalence among individuals with unexplained developmental disorders is estimated at 4.2:10,000 [Kaplanis et al 2020].
Genetically Related (Allelic) Disorders
Truncating variants in the N-terminal third of MN1 are associated with a phenotype partly overlapping but distinct from that of MN1 C-terminal truncation (MCTT) syndrome, such as conductive hearing loss and a range of speech defects (individuals 24, 25, and 26 in Mak et al [2020]). Unlike the C-terminal pathogenic variants seen in MCTT syndrome, these variants are predicted to induce nonsense-mediated mRNA decay, and individuals with this disorder had only mild developmental delay with no intellectual disability or facial gestalt reminiscent of MCTT syndrome.
Contiguous gene deletions of various sizes containing MN1 and other adjacent genes and one instance of deletion involving only MN1 have been described in individuals with variable neurodevelopmental and facial anomalies. The most frequent findings reported in these individuals are cleft or high-arched palate, micro- and/or retrognathia, hypertelorism, low-set and/or dysplastic ears, hypoplastic corpus callosum, mild-to-moderate developmental delay, and intellectual disability including delayed speech [Mak et al 2020]. Individuals with these contiguous deletions do not exhibit a consistent facial gestalt, and they do not resemble individuals with MCTT syndrome. Furthermore, review of brain imaging for these individuals did not reveal rhombencephalosynapsis or the other findings frequently observed in individuals with C-terminal truncating variants.
Differential Diagnosis
Gomez-Lopez-Hernandez syndrome (GLHS) (OMIM 601853). Like MN1 C-terminal truncation (MCTT) syndrome, GLHS is associated with turricephaly, midface hypoplasia, craniosynostosis, and rhombencephalosynapsis. Unlike MCTT syndrome, GLHS is also known to be associated with trigeminal anesthesia, scalp alopecia, corneal opacities, short stature, and ataxia. The genetic cause of GLHS is unknown.
Management
Evaluations Following Initial Diagnosis
To establish the extent of disease and needs in an individual diagnosed with MN1 C-terminal truncation (MCTT) syndrome, the evaluations summarized in Table 4 (if not performed as part of the evaluation that led to the diagnosis) are recommended.
Table 4.
Recommended Evaluations Following Initial Diagnosis in Individuals with MN1 C-Terminal Truncation Syndrome
Treatment of Manifestations
Table 5.
Treatment of Manifestations in Individuals with MN1 C-Terminal Truncation Syndrome
Developmental Delay / Intellectual Disability Management Issues
The following information represents typical management recommendations for individuals with developmental delay / intellectual disability in the United States; standard recommendations may vary from country to country.
Ages 0-3 years. Referral to an early intervention program is recommended for access to occupational, physical, speech, and feeding therapy as well as infant mental health services, special educators, and sensory impairment specialists. In the US, early intervention is a federally funded program available in all states that provides in-home services to target individual therapy needs.
Ages 3-5 years. In the US, developmental preschool through the local public school district is recommended. Before placement, an evaluation is made to determine needed services and therapies and an individualized education plan (IEP) is developed for those who qualify based on established motor, language, social, or cognitive delay. The early intervention program typically assists with this transition. Developmental preschool is center based; for children too medically unstable to attend, home-based services are provided.
All ages. Consultation with a developmental pediatrician is recommended to ensure the involvement of appropriate community, state, and educational agencies (US) and to support parents in maximizing quality of life. Some issues to consider:
- IEP services:
- An IEP provides specially designed instruction and related services to children who qualify.
- IEP services will be reviewed annually to determine whether any changes are needed.
- Special education law requires that children participating in an IEP be in the least restrictive environment feasible at school and included in general education as much as possible, when and where appropriate.
- Vision and hearing consultants should be a part of the child's IEP team to support access to academic material.
- PT, OT, and speech services will be provided in the IEP to the extent that the need affects the child's access to academic material. Beyond that, private supportive therapies based on the affected individual's needs may be considered. Specific recommendations regarding type of therapy can be made by a developmental pediatrician.
- As a child enters the teen years, a transition plan should be discussed and incorporated in the IEP. For those receiving IEP services, the public school district is required to provide services until age 21.
- A 504 plan (Section 504: a US federal statute that prohibits discrimination based on disability) can be considered for those who require accommodations or modifications such as front-of-class seating, assistive technology devices, classroom scribes, extra time between classes, modified assignments, and enlarged text.
- Developmental Disabilities Administration (DDA) enrollment is recommended. DDA is a US public agency that provides services and support to qualified individuals. Eligibility differs by state but is typically determined by diagnosis and/or associated cognitive/adaptive disabilities.
- Families with limited income and resources may also qualify for supplemental security income (SSI) for their child with a disability.
Motor Dysfunction
Gross motor dysfunction
- Physical therapy is recommended to maximize mobility and to reduce the risk for later-onset orthopedic complications (e.g., contractures, scoliosis, hip dislocation).
- Consider use of durable medical equipment and positioning devices as needed (e.g., wheelchairs, walkers, bath chairs, orthotics, adaptive strollers).
Fine motor dysfunction. Occupational therapy is recommended for difficulty with fine motor skills that affect adaptive function such as feeding, grooming, dressing, and writing.
Oral motor dysfunction should be assessed at each visit and clinical feeding evaluations and/or radiographic swallowing studies should be obtained for choking/gagging during feeds, poor weight gain, frequent respiratory illnesses, or feeding refusal that is not otherwise explained. Assuming that the child is safe to eat by mouth, feeding therapy (typically from an occupational or speech therapist) is recommended to help improve coordination or sensory-related feeding issues. Feeds can be thickened or chilled for safety. When feeding dysfunction is severe, an NG-tube or G-tube may be necessary.
Communication issues. Consider evaluation for alternative means of communication (e.g., augmentative and alternative communication [AAC]) for individuals who have expressive language difficulties. An AAC evaluation can be completed by a speech-language pathologist who has expertise in the area. The evaluation will consider cognitive abilities and sensory impairments to determine the most appropriate form of communication. AAC devices can range from low-tech, such as picture exchange communication, to high-tech, such as voice-generating devices. Contrary to popular belief, AAC devices do not hinder verbal development of speech, but rather support optimal speech and language development.
Social/Behavioral Concerns
Children may qualify for and benefit from interventions used in treatment of autism spectrum disorder, including applied behavior analysis (ABA). ABA therapy is targeted to the individual child's behavioral, social, and adaptive strengths and weaknesses and typically performed one on one with a board-certified behavior analyst.
Consultation with a developmental pediatrician may be helpful in guiding parents through appropriate behavior management strategies or providing prescription medications, such as medication used to treat attention-deficit/hyperactivity disorder, when necessary.
Concerns about serious aggressive, self-injurious, or destructive behavior can be addressed by a pediatric psychiatrist, developmental pediatrician, or psychologist with particular interest in management of these types of behaviors.
Surveillance
Table 6.
Recommended Surveillance for Individuals with MN1 C-Terminal Truncation Syndrome
Evaluation of Relatives at Risk
See Genetic Counseling for issues related to testing of at-risk relatives for genetic counseling purposes.
Therapies Under Investigation
Search ClinicalTrials.gov in the US and EU Clinical Trials Register in Europe for access to information on clinical studies for a wide range of diseases and conditions. Note: There may not be clinical trials for this disorder.
Genetic Counseling
Genetic counseling is the process of providing individuals and families with information on the nature, mode(s) of inheritance, and implications of genetic disorders to help them make informed medical and personal decisions. The following section deals with genetic risk assessment and the use of family history and genetic testing to clarify genetic status for family members; it is not meant to address all personal, cultural, or ethical issues that may arise or to substitute for consultation with a genetics professional. —ED.
Mode of Inheritance
MN1 C-terminal truncation (MCTT) syndrome is an autosomal dominant disorder typically caused by a de novo pathogenic variant.
Risk to Family Members
Parents of a proband
- Molecular genetic testing is recommended to confirm the genetic status of the parents and to allow reliable recurrence risk assessment.
- If the pathogenic variant identified in the proband is not identified in either parent, the following possibilities should be considered:
- The proband has a de novo pathogenic variant. Note: A pathogenic variant is reported as "de novo" if: (1) the pathogenic variant found in the proband is not detected in parental DNA; and (2) parental identity testing has confirmed biological maternity and paternity. If parental identity testing is not performed, the variant is reported as "assumed de novo" [Richards et al 2015].
- The proband inherited a pathogenic variant from a parent with germline (or somatic and germline) mosaicism.* Note: Testing of parental leukocyte DNA may not detect all instances of somatic mosaicism.* A parent with somatic and germline mosaicism for an MN1 pathogenic variant may be mildly/minimally affected. In one family, the mosaic father of affected sibs had mild/nondistinctive features of MCTT syndrome (i.e., mildly dysplastic ears and a high, narrow palate) [Mak et al 2020].
Sibs of a proband. The risk to the sibs of the proband depends on the genetic status of the proband's parents:
- If a parent of the proband has the MN1 pathogenic variant, the risk to the sibs of inheriting the variant is 50%.
- If the MN1 pathogenic variant identified in the proband cannot be detected in the leukocyte DNA of either parent, the recurrence risk to sibs is slightly greater than that of the general population because of the possibility of parental somatic and/or germline mosaicism [Mak et al 2020].
Offspring of a proband
- The majority of probands reported to date with MCTT syndrome whose parents have undergone molecular genetic testing have the disorder as a result of a de novo MN1 pathogenic variant.
- Each child of an individual with MCTT syndrome has a 50% chance of inheriting the MN1 pathogenic variant.
- Individuals with MCTT syndrome are not known to reproduce; however, many are not yet of reproductive age.
Other family members. Given that most probands with MCTT syndrome reported to date have the disorder as a result of a de novo MN1 pathogenic variant, the risk to other family members is presumed to be low.
Related Genetic Counseling Issues
Family planning
- The optimal time for determination of genetic risk and discussion of the availability of prenatal/preimplantation genetic testing is before pregnancy.
- It is appropriate to offer genetic counseling (including discussion of potential risks to offspring and reproductive options) to parents of affected individuals.
Prenatal Testing and Preimplantation Genetic Testing
Once the MN1 pathogenic variant has been identified in an affected family member, prenatal/preimplantation genetic testing are possible. Risk to future pregnancies is presumed to be low as the proband most likely has a de novo MN1 pathogenic variant. However, risk to future pregnancies is slightly greater than that of the general population because of the possibility of parental germline mosaicism [Mak et al 2020].
Differences in perspective may exist among medical professionals and within families regarding the use of prenatal testing. While most centers would consider use of prenatal testing to be a personal decision, discussion of these issues may be helpful.
Resources
GeneReviews staff has selected the following disease-specific and/or umbrella support organizations and/or registries for the benefit of individuals with this disorder and their families. GeneReviews is not responsible for the information provided by other organizations. For information on selection criteria, click here.
- Human Disease Genes Website Series - RegistryThis website was created to share and collect information about clinic, management and research projects to gather more knowledge and provide better treatment of patients with mutations in the MN1 gene.
Molecular Genetics
Information in the Molecular Genetics and OMIM tables may differ from that elsewhere in the GeneReview: tables may contain more recent information. —ED.
Table A.
MN1 C-Terminal Truncation Syndrome: Genes and Databases
Table B.
OMIM Entries for MN1 C-Terminal Truncation Syndrome (View All in OMIM)
Molecular Pathogenesis
MN1, a two-exon gene, encodes a transcriptional coregulator that is predicted to be involved in transcriptional regulation of target genes through interaction with the transcription factors PBX1, PKNOX1, and ZBTB24.
All MN1 C-terminal truncation (MCTT) syndrome-causing variants fall in the terminal exon, exon 2, or the extreme 3' region of exon 1. These late-truncating variants are predicted to escape nonsense-mediated mRNA decay and produce a truncated protein [Mak et al 2020]. An in vitro study demonstrated that a C-terminal region deletion also led to increased protein stability, diminished cell proliferation, and enhanced MN1 aggregation in nuclei, consistent with a gain-of-function disease mechanism [Miyake et al 2020].
Truncated MN1 impairs the binding of the E3 ubiquitin ligase RING1; therefore, C-terminal deletion may interfere with the interaction of MN1 with molecules related to the ubiquitin-mediated proteasome pathway, increasing the amount of abnormal protein. This increase would lead to the dysregulation of MN1 target genes [Miyake et al 2020].
Mechanism of disease causation. Likely gain of function mediated by expression of truncated MN1 protein
Table 7.
Notable MN1 Pathogenic Variants
Cancer and Benign Tumors
Fusion of MN1 is implicated in somatic variants observed in leukemic cells [Shao et al 2018] and neuromalignancies [Burford et al 2018]. However, cancer has not been reported in any individuals with MCTT syndrome to date.
Chapter Notes
Author Notes
Author's website: paed.hku.hk/menu/staff/bhychung/bhychung.html
Dr Brian Hon-Yin Chung is Clinical Associate Professor / Honorary Consultant in the Department of Pædiatrics and Adolescent Medicine and the Department of Obstetrics & Gynæcology of the University of Hong Kong. Dr Chung takes care of patients and families with genetic disorders. A clinical geneticist and academic paediatrician by training, he focuses on accurate delineation of the clinical phenotype/natural history of genetic syndromes and how genetic and epigenetic factors contribute to disease susceptibility, using state-of-the-art genomic technologies.
Acknowledgments
We are extremely grateful to the families for their participation, and our collaborators for their valuable contribution to our knowledge about these disorders.
Revision History
- 13 August 2020 (bp) Review posted live
- 22 April 2020 (bc) Original submission
References
Literature Cited
- Burford A, Mackay A, Popov S, Vinci M, Carvalho D, Clarke M, Izquierdo E, Avery A, Jacques TS, Ingram WJ, Moore AS. The ten-year evolutionary trajectory of a highly recurrent paediatric high grade neuroepithelial tumour with MN1: BEND2 fusion. Sci Rep. 2018;8:1032. [PMC free article: PMC5773598] [PubMed: 29348602]
- Kaplanis J, Samocha KE, Wiel L, Zhang Z, Arvai KJ, Eberhardt RY, Gallone G, Lelieveld SH, Martin HC, McRae JF, Short PJ, Torene RI, de Boer E, Danecek P, Gardner EJ, Huang N, Lord J, Martincorena I, Pfundt R, Reijnders MRF, Yeung A, Yntema HG. Deciphering Developmental Disorders Study, Vissers LELM, Juusola J, Wright CF, Brunner HG, Firth HV, FitzPatrick DR, Barrett JC, Hurles ME, Gilissen C, Retterer K. Evidence for 28 genetic disorders discovered by combining healthcare and research data. Nature. 2020;586:757–62. [PMC free article: PMC7116826] [PubMed: 33057194]
- Mak CCY, Doherty D, Lin AE, Vegas N, Cho MT, Viot G, Dimartino C, Weisfeld-Adams JD, Lessel D, Joss S, Li C, Gonzaga-Jauregui C, Zarate YA, Ehmke N, Horn D, Troyer C, Kant SG, Lee Y, Ishak GE, Leung G, Barone Pritchard A, Yang S, Bend EG, Filippini F, Roadhouse C, Lebrun N, Mehaffey MG, Martin PM, Apple B, Millan F, Puk O, Hoffer MJV, Henderson LB, McGowan R, Wentzensen IM, Pei S, Zahir FR, Yu M, Gibson WT, Seman A, Steeves M, Murrell JR, Luettgen S, Francisco E, Strom TM, Amlie-Wolf L, Kaindl AM, Wilson WG, Halbach S, Basel-Salmon L, Lev-El N, Denecke J, Vissers LELM, Radtke K, Chelly J, Zackai E, Friedman JM, Bamshad MJ, Nickerson DA, Reid RR, Devriendt K, Chae JH, Stolerman E, McDougall C, Powis Z, Bienvenu T, Tan TY, Orenstein N, Dobyns WB, Shieh JT, Choi M, Waggoner D, Gripp KW, Parker MJ, Stoler J, Lyonnet S, Cormier-Daire V, Viskochil D, Hoffman TL, Amiel J, Chung BHY, Gordon CT, et al. MN1 C-terminal truncation syndrome is a novel neurodevelopmental and craniofacial disorder with partial rhombencephalosynapsis. Brain. 2020;143:55–68. [PMC free article: PMC7962909] [PubMed: 31834374]
- McRae JF, Clayton S, Fitzgerald TW, Kaplanis J, Prigmore E, Rajan D, Sifrim A, Aitken S, Akawi N, Alvi M, et al. Prevalence and architecture of de novo mutations in developmental disorders. Nature. 2017;542:433–8. [PMC free article: PMC6016744] [PubMed: 28135719]
- Miyake N, Takahashi H, Nakamura K, Isidor B, Hiraki Y, Koshimizu E, Shiina M, Sasaki K, Suzuki H, Abe R, Kimura Y, Akiyama T, Tomizawa SI, Hirose T, Hamanaka K, Miyatake S, Mitsuhashi S, Mizuguchi T, Takata A, Obo K, Kato M, Ogata K, Matsumoto N. Gain-of-function MN1 truncation variants cause a recognizable syndrome with craniofacial and brain abnormalities. Am J Hum Genet. 2020;106:13–25. [PMC free article: PMC7042485] [PubMed: 31839203]
- Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, Grody WW, Hegde M, Lyon E, Spector E, Voelkerding K, Rehm HL, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17:405–24. [PMC free article: PMC4544753] [PubMed: 25741868]
- Rossi M, El-Khechen D, Black MH, Farwell Hagman KD, Tang S, Powis Z. Outcomes of diagnostic exome sequencing in patients with diagnosed or suspected autism spectrum disorders. Pediatr Neurol. 2017;70:34–43.e2. [PubMed: 28330790]
- Shao H, Cen J, Chen S, Qiu H, Pan J. Myeloid neoplasms with t (12; 22)(p13; q12)/MN1-EVT6: a systematic review of 12 cases. Ann Hematol. 2018;97:417–24. [PubMed: 29273914]
Publication Details
Author Information and Affiliations
LKS Faculty of Medicine
University of Hong Kong
Hong Kong Special Administrative Region, China
LKS Faculty of Medicine
University of Hong Kong
Hong Kong Special Administrative Region, China
LKS Faculty of Medicine
University of Hong Kong
Hong Kong Special Administrative Region, China
Harvard Medical School
Co-Director, Turner Syndrome Program
Co-Director, Myhre Syndrome Clinic
MassGeneral Hospital for Children
Boston, Massachusetts
Institut National de la Santé et de la Recherche Médicale (INSERM)
Institut Imagine
Paris, France
Seattle Children's Hospital
Seattle, Washington
Institut National de la Santé et de la Recherche Médicale (INSERM)
Institut Imagine
Paris, France
LKS Faculty of Medicine
University of Hong Kong
Hong Kong Special Administrative Region, China
Publication History
Initial Posting: August 13, 2020.
Copyright
GeneReviews® chapters are owned by the University of Washington. Permission is hereby granted to reproduce, distribute, and translate copies of content materials for noncommercial research purposes only, provided that (i) credit for source (http://www.genereviews.org/) and copyright (© 1993-2024 University of Washington) are included with each copy; (ii) a link to the original material is provided whenever the material is published elsewhere on the Web; and (iii) reproducers, distributors, and/or translators comply with the GeneReviews® Copyright Notice and Usage Disclaimer. No further modifications are allowed. For clarity, excerpts of GeneReviews chapters for use in lab reports and clinic notes are a permitted use.
For more information, see the GeneReviews® Copyright Notice and Usage Disclaimer.
For questions regarding permissions or whether a specified use is allowed, contact: ude.wu@tssamda.
Publisher
University of Washington, Seattle, Seattle (WA)
NLM Citation
Mak CCY, Fung JLF, Lee M, et al. MN1 C-Terminal Truncation Syndrome. 2020 Aug 13. In: Adam MP, Feldman J, Mirzaa GM, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2024.