Summary
Clinical characteristics.
Osteoglophonic dysplasia (OGD) is characterized by multisuture craniosynostosis (including cloverleaf skull), distinctive craniofacial features (prominent forehead, proptosis, widely spaced eyes, low-set ears, midface retrusion, short nose, anteverted nares, prognathism, high palate, failure of tooth eruption, and gingival overgrowth), profound short stature with rhizomelia, and short, broad hands and feet. Radiographs show copper beaten appearance to skull, multiple cystic long bone lesions consistent with non-ossifying fibromas, irregular vertebral bodies, and osteopenia with increased risk of fractures.
Diagnosis/testing.
The diagnosis of OGD is established in a proband with characteristic clinical and imaging findings and a heterozygous pathogenic gain-of-function variant in FGFR1 identified by molecular genetic testing.
Management.
Treatment of manifestations: Management of musculoskeletal manifestations per skeletal dysplasia or physiatry clinic; address mobility issues in those with bone deformity; early intervention such as physical therapy, occupational therapy, and speech therapy to optimize developmental outcomes; surgical repair in the first year of life in those with multisuture craniosynostosis; early initiation of topical eye lubrication in those with inadequate lid closure; jaw surgery to advance the midface; pediatric dental and orthodontic care; surgical interventions as needed for sleep apnea; treatment with phosphate as needed per endocrinologist; standard treatment of hernias per gastroenterologist/surgeon.
Surveillance: At each visit monitor growth, developmental progress, and educational needs, and assess for recurrent and pathologic fractures, incomplete eyelid closure, and manifestations of sleep apnea. Clinical evaluation of head circumference and for manifestations of increased intracranial pressure at least every three months in the first year of life. Evaluation by a craniofacial orthodontist when secondary teeth have erupted. Assess serum phosphate and serum FGF23 as recommended by endocrinologist. Monitor body temperature following sedation for procedures.
Agents/circumstances to avoid: Sports restrictions may be necessary for activities that carry a potential for head or neck injury; individuals with severe proptosis need to wear protective eyewear during activities with risk of eye injury.
Evaluation of relatives at risk: It is appropriate to clarify the genetic status of apparently asymptomatic older and younger at-risk relatives of an affected individual in order to identify as early as possible those who would benefit from prompt initiation of treatment of developmental and craniofacial manifestations.
Genetic counseling.
OGD is inherited in an autosomal dominant manner. Most individuals diagnosed with OGD represent simplex cases; some individuals diagnosed with OGD have an affected parent. Each child of an individual with OGD has a 50% chance of inheriting the FGFR1 pathogenic variant. Once the FGFR1 pathogenic variant has been identified in an affected family member, prenatal and preimplantation genetic testing are possible. Because many individuals with short stature have reproductive partners with short stature, offspring of individuals with OGD may be at risk of having double heterozygosity for two dominantly inherited bone growth disorders; the phenotypes of these individuals are distinct from those of the parents, and the affected individuals have serious sequelae and poor outcomes. Pregnant women carrying fetuses affected by OGD should be monitored during pregnancy for features that can affect early morbidity and mortality and should be encouraged to deliver in a hospital with ready access to a pediatric otolaryngologist, plastic surgeon, neurosurgeon, and pulmonary medicine specialist.
Diagnosis
Suggestive Findings
Osteoglophonic dysplasia (OGD) should be suspected in probands with the following clinical, imaging, and laboratory findings:
Clinical findings
- Craniofacial
- Multisuture craniosynostosis (including cloverleaf skull)
- Prominent forehead
- Proptosis
- Widely spaced eyes
- Low-set ears
- Midface retrusion
- Short nose
- Anteverted nares
- Prognathism
- High palate
- Failure of tooth eruption
- Gingival overgrowth
- Skeletal
- Short stature
- Rhizomelic limb shortening
- Short, broad hands and feet
- Genu varum
- Overlapping toes
- Pathologic fractures
- Other
- Poor weight gain
- Increased body temperature
- Increased sensitivity to heat
- Excessive sweating
- Nasal obstruction
- Short neck
- Inguinal hernia
- Developmental delay (primarily speech delay)
Imaging findings (See Figure 1.)
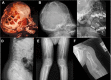
Figure 1.
Radiographs of individuals with osteoglophonic dysplasia A, B, and C. Lateral imaging of the skull showing copper beaten appearance and unerupted teeth
- Skull. Copper beaten appearance
- Teeth. Multiple unerupted permanent tooth buds in mandible and maxilla
- Long bones. Multiple cystic bone lesions consistent with non-ossifying fibromas
- Vertebral bodies. Irregular platyspondyly with anterior projection and concavity of their posterior portions
- Osteopenia
Laboratory findings
- Hypophosphatemia
- Normal to increased serum fibroblast growth factor 23 (FGF23)
Establishing the Diagnosis
The diagnosis of OGD is established in a proband with suggestive findings and a heterozygous pathogenic (or likely pathogenic) gain-of-function variant in FGFR1 identified by molecular genetic testing (see Table 1).
Note: (1) Per ACMG/AMP variant interpretation guidelines, the terms "pathogenic variant" and "likely pathogenic variant" are synonymous in a clinical setting, meaning that both are considered diagnostic and can be used for clinical decision making [Richards et al 2015]. Reference to "pathogenic variants" in this section is understood to include likely pathogenic variants. (2) Identification of a heterozygous FGFR1 variant of uncertain significance does not establish or rule out the diagnosis.
Molecular genetic testing approaches can include a combination of gene-targeted testing (single gene testing, multigene panel) and comprehensive genomic testing (exome sequencing, genome sequencing). Gene-targeted testing requires that the clinician determine which gene(s) are likely involved (see Option 1), whereas comprehensive genomic testing does not (see Option 2).
Option 1
Single-gene testing. Sequence analysis of FGFR1. Note: (1) All previously reported variants fall in the transmembrane domain or the adjacent immunoglobulin-like III domain (see Molecular Genetics) (2) OGD occurs through a gain-of-function mechanism. Only missense pathogenic variants have been reported; deletion or duplication of FGFR1 are not expected to cause this disorder.
A skeletal dysplasia multigene panel that includes FGFR1 and other genes of interest (see Differential Diagnosis) may be considered to identify the genetic cause of the condition while limiting identification of variants of uncertain significance and pathogenic variants in genes that do not explain the underlying phenotype. Note: (1) The genes included in the panel and the diagnostic sensitivity of the testing used for each gene vary by laboratory and are likely to change over time. (2) Some multigene panels may include genes not associated with the condition discussed in this GeneReview. (3) In some laboratories, panel options may include a custom laboratory-designed panel and/or custom phenotype-focused exome analysis that includes genes specified by the clinician. (4) Methods used in a panel may include sequence analysis, deletion/duplication analysis, and/or other non-sequencing-based tests.
For an introduction to multigene panels click here. More detailed information for clinicians ordering genetic tests can be found here.
Option 2
When the phenotype is indistinguishable from many other skeletal dysplasias, comprehensive genomic testing does not require the clinician to determine which gene is likely involved. Exome sequencing is most commonly used; genome sequencing is also possible. To date, all FGFR1 pathogenic variants are within the coding region and are likely to be identified on exome sequencing.
For an introduction to comprehensive genomic testing click here. More detailed information for clinicians ordering genomic testing can be found here.
Table 1.
Molecular Genetic Testing Used in Osteoglophonic Dysplasia
Clinical Characteristics
Clinical Description
Osteoglophonic dysplasia (OGD) is a skeletal dysplasia characterized by multisuture craniosynostosis (including premature fusion of the coronal, sagittal, lambdoid, and metopic sutures), distinctive craniofacial features, unerupted teeth, profound short stature, and multiple cystic bone lesions consistent with non-ossifying fibromas. To date, 24 individuals with OGD from 19 families have been reported and/or identified; of these, 14 individuals have had molecular genetic testing with a pathogenic variant identified in FGFR1 [Marzin et al 2020, Zou et al 2022]. The remaining ten individuals did not have molecular testing. The following description of the phenotypic features associated with this condition is based on these reports.
Table 2.
Osteoglophonic Dysplasia: Frequency of Select Features
Skeletal. Multiple cystic lesions consistent with non-ossifying fibromas are seen on radiologic images of the proximal and distal femur, distal tibia and fibula, iliac bones, proximal humerus, and distal radius and ulna (Figure 1E, 1F). The cystic lesions appear early in life and gradually increase in size and number during childhood; later, they gradually ossify, regress, or disappear after skeletal maturity. The tubular bones appear broad and short with marked dysplastic changes at the epiphyseal ossification centers. Spinal imaging shows platyspondyly with anterior beaking and posterior scalloping of the lower thoracic and lumbar vertebral bodies (Figure 1D) [Kelley et al 1983, Azouz & Kozlowski 1997, Sargar et al 2017, Marzin et al 2020]. Other skeletal features include rhizomelic limb shortening, short, broad hands and feet, genu varum, pseudoarthrosis, pathologic fractures, and overlapping toes, where the 3rd toe is overlapping or underriding the 2nd and 4th toes [A Othman, H Babcock, & C Ferreira, personal observations].
Growth. Individuals with OGD show impaired postnatal growth. Affected infants are below the 3rd centile for length but profound short stature becomes more evident with age. Rhizomelic limb shortening becomes increasingly apparent in childhood. Milder short stature has been reported in some individuals with normal to low normal height; adult height ranges between 97 and 154 cm [Beighton 1989, Marzin et al 2020].
Poor weight gain can be attributed to feeding difficulties, choanal atresia, or nasal obstruction with airway and breathing problems, which can occur during infancy as a result of craniofacial abnormalities and may rarely lead to death [Santos et al 1988].
Craniofacial. Multisuture craniosynostosis is present in most individuals. Head shape depends on the sutures involved and the timing of premature fusion, ranging from normal head shape to turribrachycephaly. Individuals without craniosynostosis have been described [Marzin et al 2020]. Other craniofacial features, such as a prominent forehead, midface retrusion, maxillary hypoplasia, prognathism, and proptosis, are evident at birth. Widely spaced eyes, low-set ears, short nose, anteverted nares, high palate, and gingival overgrowth are other early features. Choanal atresia or nasal obstruction contributes to feeding difficulties, airway and breathing problems, and poor weight gain during infancy and may rarely lead to death [Santos et al 1988]. Craniofacial abnormalities such as midface retrusion and maxillary hypoplasia can contribute to multilevel airway obstruction and may result in obstructive sleep apnea.
Delayed primary and secondary teeth eruption is a common feature during childhood. Skull radiographs typically show impacted permanent tooth buds, with a characteristic copper beaten appearance that may regress by adulthood [Kuthiroly et al 2017] (Figure 1A-C). Giant cell granuloma of the jaw has also been reported.
Endocrine. Fibroblast growth factor 23 (FGF23) serum levels may increase over time, leading to renal phosphate wasting. Decreased phosphate absorption in the kidneys can result in hypophosphatemia, decreased bone mineralization (with increased risk of fractures), and disturbed vitamin D metabolism. Phosphorus supplementation can be beneficial in those with elevated FGF23-mediated hypophosphatemia.
Neurodevelopment. Development, particularly speech development, can be delayed in early childhood but improves with age. Intelligence is normal unless hydrocephalus or other central nervous system complications occur. For children with multisuture craniosynostosis, early and aggressive surgical intervention to address increased intracranial pressure may prevent intellectual disability. Motor limitation can manifest during childhood as a result of severe cystic bone lesions and osteopenia, leading to bone pain, bone fractures, and skeletal deformities [Kumar et al 2021; A Othman, H Babcock, & C Ferreira, personal observations].
Gastrointestinal. Inguinal hernia and pyloric stenosis have each been reported in two individuals [Kelley et al 1983, Beighton 1989, Holder et al 2017].
Temperature / heat intolerance. Increased body temperature and sensitivity to heat accompanied by excessive sweating has been reported in two individuals [A Othman, H Babcock, & C Ferreira, personal observations].
Genotype-Phenotype Correlations
No genotype-phenotype correlations have been identified.
Penetrance
Penetrance is complete.
Nomenclature
The term osteoglophonic is derived from the Greek word meaning "hollowed out" and refers to the characteristic multiple non-ossifying fibromas that appear as cystic radiolucent lesions on radiologic images.
This phenotype is thought to have been first described by Sir Thomas Fairbank [Fairbank 1951], who named it "acrocephaly with abnormalities of the extremities," and then by Theodore E Keats [Keats et al 1975] who named it "craniofacial dysostosis with fibrous metaphyseal defects." The syndrome was then referred to as "Fairbank-Keats syndrome" before it was formally described as "osteoglophonic dwarfism" and finally "osteoglophonic dysplasia" by Peter Beighton [Beighton et al 1980, Beighton 1989].
In the 2023 revision of the Nosology of Genetic Skeletal Disorders [Unger et al 2023], osteoglophonic dysplasia is referred to as FGFR1-related osteoglophonic dysplasia and included in the disorganized development of skeletal components group.
Prevalence
OGD is rare, with 22 individuals (from 17 different families) reported to date [Fairbank 1951, Keats et al 1975, Beighton et al 1980, Kelley et al 1983, Santos et al 1988, Sklower Brooks et al 1996, Azouz & Kozlowski 1997, White et al 2005, Farrow et al 2006, Shankar et al 2010, Sow et al 2010, Holder et al 2017, Kuthiroly et al 2017, Marzin et al 2020, Zou et al 2022]. Two additional unrelated individuals are known to the authors. There is no known geographic predilection.
Genetically Related (Allelic) Disorders
Germline pathogenic variants in FGFR1 are known to be associated with a wide spectrum of phenotypes (see Table 3).
Table 3.
FGFR1 Allelic Disorders
Mosaic activating pathogenic variants in FGFR1 are associated with encephalocraniocutaneous lipomatosis (ECCL). The pathogenic variants reported in ECCL are of postzygotic origin but arise early during development. ECCL comprises a spectrum of predominantly congenital anomalies. In its typical form, ECCL is characterized by congenital skin, eye, and brain anomalies, in particular intracranial and spinal lipomas.
Differential Diagnosis
Table 4.
Genes of Interest in the Differential Diagnosis of Osteoglophonic Dysplasia
Management
No clinical practice guidelines for osteoglophonic dysplasia (OGD) have been published.
Evaluations Following Initial Diagnosis
To establish the extent of disease and needs in an individual diagnosed with OGD, the evaluations summarized in Table 5 (if not performed as part of the evaluation that led to the diagnosis) are recommended.
Table 5.
Osteoglophonic Dysplasia: Recommended Evaluations Following Initial Diagnosis
Treatment of Manifestations
There is no cure for OGD. Supportive care to improve quality of life, maximize function, and reduce complications is recommended. This ideally involves multidisciplinary care by specialists in relevant fields (see Table 6).
Table 6.
Osteoglophonic Dysplasia: Treatment of Manifestations
Surveillance
To monitor existing manifestations, the individual's response to supportive care, and the emergence of new manifestations, the evaluations summarized in Table 7 are recommended.
Table 7.
Osteoglophonic Dysplasia: Recommended Surveillance
Agents/Circumstances to Avoid
Individuals with OGD may require sports restrictions for activities that carry a potential for head or neck injury.
Individuals with severe proptosis need to wear protective eyewear during activities with risk of eye injury (e.g., ball sports).
Evaluation of Relatives at Risk
It is appropriate to clarify the genetic status of apparently asymptomatic older and younger at-risk relatives of an affected individual in order to identify as early as possible those who would benefit from prompt initiation of treatment of developmental and craniofacial manifestations.
See Genetic Counseling for issues related to testing of at-risk relatives for genetic counseling purposes.
Pregnancy Management
No published studies address management of pregnancy in women with OGD.
See MotherToBaby for further information on medication use during pregnancy.
Therapies Under Investigation
Search ClinicalTrials.gov in the US and EU Clinical Trials Register in Europe for access to information on clinical studies for a wide range of diseases and conditions. Note: There may not be clinical trials for this disorder.
Genetic Counseling
Genetic counseling is the process of providing individuals and families with information on the nature, mode(s) of inheritance, and implications of genetic disorders to help them make informed medical and personal decisions. The following section deals with genetic risk assessment and the use of family history and genetic testing to clarify genetic status for family members; it is not meant to address all personal, cultural, or ethical issues that may arise or to substitute for consultation with a genetics professional. —ED.
Mode of Inheritance
Osteoglophonic dysplasia (OGD) is inherited in an autosomal dominant manner.
Risk to Family Members
Parents of a proband
- Most individuals diagnosed with OGD represent simplex cases (i.e., the only family member known to be affected).
- Some individuals diagnosed with OGD have an affected parent. Of the 19 families reported and/or identified to date, parent-to-child transmission of an FGFR1 pathogenic variant has been reported in four families.
- If a molecular diagnosis has been established in the proband and the proband appears to be the only affected family member, molecular genetic testing is recommended for the parents of the proband to evaluate their genetic status and inform recurrence risk assessment.
- If the pathogenic variant identified in the proband is not identified in either parent and parental identity testing has confirmed biological maternity and paternity, the following possibilities should be considered:
- The proband has a de novo pathogenic variant.
- The proband inherited a pathogenic variant from a parent with germline (or somatic and germline) mosaicism. Note: Testing of parental leukocyte DNA may not detect all instances of somatic mosaicism and will not detect a pathogenic variant that is present in the germ (gonadal) cells only.
Sibs of a proband. The risk to the sibs of the proband depends on the genetic status of the proband's parents:
- If a parent of the proband is affected and/or is known to have the pathogenic variant identified in the proband, the risk to the sibs of inheriting the pathogenic variant is 50%.
- If a molecular diagnosis has been established in the proband and the FGFR1 pathogenic variant identified in the proband cannot be detected in the leukocyte DNA of either parent, the recurrence risk to sibs is estimated to be 1% because of the possibility (general, not specific to this condition) of parental germline mosaicism [Rahbari et al 2016].
- If the parents have not been tested for the FGFR1 pathogenic variant but are clinically unaffected, the risk to the sibs of a proband appears to be low. However, sibs of a proband with clinically unaffected parents are still presumed to be at increased risk for OGD because of the possibility of parental germline mosaicism.
Offspring of a proband
- Each child of an individual with OGD has a 50% chance of inheriting the FGFR1 pathogenic variant.
- Because many individuals with short stature have reproductive partners with short stature, offspring of individuals with OGD may be at risk of having double heterozygosity for two dominantly inherited bone growth disorders. The phenotypes of these individuals are distinct from those of the parents, and the affected individuals have serious sequelae and poor outcomes [Flynn & Pauli 2003].
Other family members. The risk to other family members depends on the status of the proband's parents: if a parent has the FGFR1 pathogenic variant, the parent's family members may be at risk.
Related Genetic Counseling Issues
Family planning
- The optimal time for determination of genetic risk and discussion of the availability of prenatal/preimplantation genetic testing is before pregnancy.
- It is appropriate to offer genetic counseling (including discussion of potential risks to offspring and reproductive options) to young adults who are affected.
Prenatal Testing and Preimplantation Genetic Testing
Once the FGFR1 pathogenic variant has been identified in an affected family member, prenatal and preimplantation genetic testing are possible.
Pregnant women carrying fetuses affected by OGD should be monitored during pregnancy for features that can affect early morbidity and mortality (including craniosynostosis and respiratory obstructions) and should be encouraged to deliver in a hospital with ready access to a pediatric otolaryngologist, plastic surgeon, neurosurgeon, and pulmonary medicine specialist.
Differences in perspective may exist among medical professionals and within families regarding the use of prenatal testing. While most centers would consider use of prenatal testing to be a personal decision, discussion of these issues may be helpful.
Resources
GeneReviews staff has selected the following disease-specific and/or umbrella support organizations and/or registries for the benefit of individuals with this disorder and their families. GeneReviews is not responsible for the information provided by other organizations. For information on selection criteria, click here.
- MedlinePlus
- American Cleft Palate-Craniofacial AssociationPhone: 919-933-9044
- Born a Hero
- Children's Craniofacial AssociationPhone: 800-535-3643Email: [email protected]
- Face Equality InternationalUnited Kingdom
- National Organization for Rare Disorders (NORD)Phone: 800-999-6673
Molecular Genetics
Information in the Molecular Genetics and OMIM tables may differ from that elsewhere in the GeneReview: tables may contain more recent information. —ED.
Table A.
Osteoglophonic Dysplasia: Genes and Databases
Table B.
OMIM Entries for Osteoglophonic Dysplasia (View All in OMIM)
Molecular Pathogenesis
FGFR1 encodes fibroblast growth factor receptor 1 (FGFR1), a fibroblast growth factor receptor protein crucial for the osteogenesis of the axial and craniofacial skeleton. FGFR1 consists of three extracellular immunoglobulin (Ig)-like domains (Ig l, Ig ll, and Ig lIl), a transmembrane (TM) domain, and an intracellular tyrosine kinase domain. Osteoglophonic dysplasia is caused by distinct heterozygous FGFR1 pathogenic gain-of-function variants affecting the TM domain or the adjacent Ig III domain; currently five causative variants are known (see Table 8). The overactivation of FGFR1 leads to excess bony fibroblast growth factor 23 (FGF23) secretion, resulting in renal phosphate wasting and hypophosphatemia.
Mechanism of disease causation. Gain of function
Table 8.
FGFR1 Pathogenic Variants Referenced in This GeneReview
Chapter Notes
Author Notes
Dr Amna A Othman ([email protected]), Ms Holly E Babcock ([email protected]), and Dr Carlos R Ferreira ([email protected]) are actively involved in clinical research regarding individuals with osteglophonic dysplasia (OGD). They would be happy to communicate with persons who have any questions regarding the diagnosis of OGD or other considerations.
Dr Carlos Ferreira is interested in hearing from clinicians treating families affected by skeletal conditions in whom no causative variant has been identified through molecular genetic testing of the genes known to be involved in this group of disorders.
Contact Dr Carlos Ferreira to inquire about review of FGFR1 variants of uncertain significance.
Revision History
- 18 April 2024 (sw) Review posted live
- 27 December 2023 (cf) Original submission
Note: Pursuant to 17 USC Section 105 of the United States Copyright Act, the GeneReview "Osteoglophonic Dysplasia" is in the public domain in the United States of America.
References
Literature Cited
- Azouz EM, Kozlowski K. Osteoglophonic dysplasia: appearance and progression of multiple nonossifying fibromata. Pediatr Radiol. 1997;27:75-8. [PubMed: 8995175]
- Beighton P. Osteoglophonic dysplasia. J Med Genet. 1989;26:572-6. [PMC free article: PMC1015696] [PubMed: 2810341]
- Beighton P, Cremin BJ, Kozlowski K. Osteoglophonic dwarfism. Pediatr Radiol. 1980;10:46-50. [PubMed: 7422392]
- Fairbank T. An Atlas of General Affections of the Skeleton. 1st ed. Edinburgh, Scotland: E & S Livingstone Ltd; 1951.
- Farrow EG, Davis SI, Mooney SD, Beighton P, Mascarenhas L, Gutierrez YR, Pitukcheewanont P, White KE. Extended mutational analyses of FGFR1 in osteoglophonic dysplasia. Am J Med Genet A. 2006;140:537-9. [PubMed: 16470795]
- Flynn MA, Pauli RM. Double heterozygosity in bone growth disorders: four new observations and review. Am J Med Genet A. 2003;121A:193-208. [PubMed: 12923858]
- Holder J, Zinn D, Samin A. Adult-onset idiopathic hypertrophic pyloric stenosis associated with osteoglophonic dysplasia and HIV: case report and review of literature. Ultrasound Q. 2017;33:77-81. [PubMed: 27599310]
- Jarzabek K, Wolczynski S, Lesniewicz R, Plessis G, Kottler ML. Evidence that FGFR1 loss-of-function mutations may cause variable skeletal malformations in patients with Kallmann syndrome. Adv Med Sci. 2012;57:314–21. [PubMed: 23154428]
- Keats TE, Smith TH, Sweet DE. Craniofacial dysotosis with fibrous metaphyseal deffects. Am J Roentgenol Radium Ther Nucl Med. 1975;124:271-5. [PubMed: 1137039]
- Kelley RI, Borns PF, Nichols D, Zackai EH. Osteoglophonic dwarfism in two generations. J Med Genet. 1983;20:436-40. [PMC free article: PMC1049176] [PubMed: 6606709]
- Kumar A, Chong YT, Jamil K, Rusli E. Severe presentation of non-ossifying fibroma of the femur in osteoglophonic dysplasia. BMJ Case Rep. 2021;14:e245415. [PMC free article: PMC8573633] [PubMed: 34740908]
- Kuthiroly S, Yesodharan D, Ghosh A, White KE, Nampoothiri S. Osteoglophonic dysplasia: phenotypic and radiological clues. J Pediatr Genet. 2017;6:247-51. [PMC free article: PMC5687321] [PubMed: 29147600]
- Marzin P, Baujat G, Gensburger D, Huber C, Bole C, Panuel M, Finidori G, De la Dure M, Cormier-Daire V. Heterozygous FGFR1 mutation may be responsible for an incomplete form of osteoglophonic dysplasia, characterized only by radiolucent bone lesions and teeth retentions. Eur J Med Genet. 2020;63:103729. [PubMed: 31319224]
- Rahbari R, Wuster A, Lindsay SJ, Hardwick RJ, Alexandrov LB, Turki SA, Dominiczak A, Morris A, Porteous D, Smith B, Stratton MR, Hurles ME, et al. Timing, rates and spectra of human germline mutation. Nat Genet. 2016;48:126-33. [PMC free article: PMC4731925] [PubMed: 26656846]
- Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, Grody WW, Hegde M, Lyon E, Spector E, Voelkerding K, Rehm HL, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17:405-24. [PMC free article: PMC4544753] [PubMed: 25741868]
- Santos H, Campos P, Alves R, Torrado A. Osteoglophonic dysplasia: a new case. Eur J Pediatr. 1988;147:547-9. [PubMed: 3409933]
- Sarfati J, Bouvattier C, Bry-Gauillard H, Cartes A, Bouligand J, Young J. Kallmann syndrome with FGFR1 and KAL1 mutations detected during fetal life. Orphanet J Rare Dis. 2015;10:71. [PMC free article: PMC4469106] [PubMed: 26051373]
- Sargar KM, Singh AK, Kao SC. Imaging of skeletal disorders caused by fibroblast growth factor receptor gene mutations. Radiographics. 2017;37:1813-30. [PubMed: 29019756]
- Shankar VN, Ajila V, Kumar G. Osteoglophonic dysplasia: a case report. J Oral Sci. 2010;52:167-71. [PubMed: 20339250]
- Sklower Brooks S, Kassner G, Qazi Q, Keogh MJ, Gorlin RJ. Osteoglophonic dysplasia: review and further delineation of the syndrome. Am J Med Genet. 1996;66:154-62. [PubMed: 8958322]
- Sow AJ, Ramli R, Latiff ZA, Ichikawa S, Gray AK, Nordin R, Abd Jabar MN, Primuharsa Putra SH, Siar CH, Econs MJ. Osteoglophonic dysplasia: a "common" mutation in a rare disease. Clin Genet. 2010;78:197-8. [PMC free article: PMC4201914] [PubMed: 20236123]
- Unger S, Ferreira CR, Mortier GR, Ali H, Bertola DR, Calder A, Cohn DH, Cormier-Daire V, Girisha KM, Hall C, Krakow D, Makitie O, Mundlos S, Nishimura G, Robertson SP, Savarirayan R, Sillence D, Simon M, Sutton VR, Warman ML, Superti-Furga A. Nosology of genetic skeletal disorders: 2023 revision. Am J Med Genet A. 2023;191:1164-209. [PMC free article: PMC10081954] [PubMed: 36779427]
- Villanueva C, de Roux N. FGFR1 mutations in Kallmann syndrome. Front Horm Res. 2010;39:51–61 [PubMed: 20389085]
- Villanueva C, Jacobson-Dickman E, Xu C, Manouvrier S, Dwyer AA, Sykiotis GP, Beenken A, Liu Y, Tommiska J, Hu Y, Tiosano D, Gerard M, Leger J, Drouin-Garraud V, Lefebvre H, Polak M, Carel JC, Phan-Hug F, Hauschild M, Plummer L, Rey JP, Raivio T, Bouloux P, Sidis Y, Mohammadi M, de Roux N, Pitteloud N. Congenital hypogonadotropic hypogonadism with split hand/foot malformation: a clinical entity with a high frequency of FGFR1 mutations. Genet Med. 2015;17:651–9. [PMC free article: PMC4430466] [PubMed: 25394172]
- Vizeneux A, Hilfiger A, Bouligand J, Pouillot M, Brailly-Tabard S, Bashamboo A, McElreavey K, Brauner R. Congenital hypogonadotropic hypogonadism during childhood: presentation and genetic analyses in 46 boys. PLoS One. 2013;8:e77827 [PMC free article: PMC3812007] [PubMed: 24204987]
- White KE, Cabral JM, Davis SI, Fishburn T, Evans WE, Ichikawa S, Fields J, Yu X, Shaw NJ, McLellan NJ, McKeown C, Fitzpatrick D, Yu K, Ornitz DM, Econs MJ. Mutations that cause osteoglophonic dysplasia define novel roles for FGFR1 in bone elongation. Am J Hum Genet. 2005;76:361-7. [PMC free article: PMC1196382] [PubMed: 15625620]
- Zou Y, Lin H, Chen W, Chang L, Cai S, Lu YG, Xu L. Abnormal eruption of teeth in relation to FGFR1 heterozygote mutation: a rare case of osteoglophonic dysplasia with 4-year follow-up. BMC Oral Health. 2022;22:36. [PMC free article: PMC8832749] [PubMed: 35148738]
Publication Details
Author Information and Affiliations
National Institutes of Health
Bethesda, Maryland
National Institutes of Health
Bethesda, Maryland
National Institutes of Health
Bethesda, Maryland
Publication History
Initial Posting: April 18, 2024.
Copyright
GeneReviews® chapters are owned by the University of Washington. Permission is hereby granted to reproduce, distribute, and translate copies of content materials for noncommercial research purposes only, provided that (i) credit for source (http://www.genereviews.org/) and copyright (© 1993-2024 University of Washington) are included with each copy; (ii) a link to the original material is provided whenever the material is published elsewhere on the Web; and (iii) reproducers, distributors, and/or translators comply with the GeneReviews® Copyright Notice and Usage Disclaimer. No further modifications are allowed. For clarity, excerpts of GeneReviews chapters for use in lab reports and clinic notes are a permitted use.
For more information, see the GeneReviews® Copyright Notice and Usage Disclaimer.
For questions regarding permissions or whether a specified use is allowed, contact: ude.wu@tssamda.
Publisher
University of Washington, Seattle, Seattle (WA)
NLM Citation
Othman AA, Babcock HE, Ferreira CR. Osteoglophonic Dysplasia. 2024 Apr 18. In: Adam MP, Feldman J, Mirzaa GM, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2024.
