Domains can be thought of as distinct functional and/or structural units of a
protein. These two classifications coincide rather often, as a matter of fact, and what
is found as an independently folding unit of a polypeptide chain also
carries specific function. Domains are often identified as recurring
(sequence or structure) units, which may exist in various
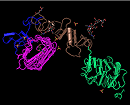
contexts. The image below illustrates four "domains" identified as structural
units in the MMDB-entry
1IGR,
chain A, as segments colored in magenta, blue, brown, and green.
In molecular evolution such domains may have been utilized as building blocks,
and may have been recombined in different arrangements to modulate protein function. We define conserved domains as recurring units in molecular evolution, the extents of which can be determined by sequence and structure analysis.
Conserved domains contain conserved sequence patterns or motifs, which
allow for their detection in polypeptide sequences. The distinction between
domains and motifs is not sharp, however, especially in the case of
short repetitive units. Functional motifs are also present outside the
scope of structurally conserved domains. The CD database is not
meant to systematically collect such motifs.

|
For this query sequence, a good correspondence exists between structural units (3D domains), identified by purely geometric criteria, and units asserted to be evolutionary conserved (domain families). The region annotated as "FU" (furin-repeat like)
overlaps with a domain-split that was suggested by the MMDB domain parser.
Click anywhere on the image to open the complete, interactive 3D record for this protein structure (1IGR) in Cn3D, a free helper application available for Windows, Macintosh, and Unix platforms. Note: Cn3D must be present on your computer, however, in order for the link to work. Cn3D installation takes only a couple of minutes and a tutorial describes the program's features and functions.
Open the 1IGR structure summary record in the Molecular Modeling Database (MMDB) to access more information about the protein, its conserved domains, and ligands (small molecules). On that page, follow the "show annotation" link to open an interactive graphic in which you can click on a conserved domain or ligand of interest to view its complete information in the Conserved Domain Database or PubChem, respectively. Click on the grey bar representing the overall protein, or on a colored bar representing an individual 3D domain of interest, to retrieve similar 3D structures, as identified by the Vector Alignment Search Tool (VAST).
View the CD-Search help document for more details about the program that was used to identify the conserved domains in the protein chain. The concise display of the conserved domains is shown here and includes specific hits, superfamilies, and multi-domains. (Open the current, interactive CD-Search results for this protein to view alignments of its sequence to a conserved domain's consensus sequence, and/or to
access a full display of all domain models found.)
|
The two types of domains shown in the 1IGR illustration above -- 3D domains and conserved domains (or "domain families") -- often coincide with each other. However, because they represent two distinct types of data -- 3D structures and protein sequences, respectively -- they reside in two distinct databases: the Entrez Structure (Molecular Modeling Database, MMDB) and the Conserved Domain Database (CDD). The former includes the spatial (X,Y,Z) coordinates of each atom in a structure (where 3D domains are identified algorithmically), while the latter shows the span and composition of a conserved protein sequence region.
Specifically, conserved domain models are based on multiple sequence alignments of related proteins spanning a variety of organisms to reveal sequence regions containing the same, or similar, patterns of amino acids. The illustration below provides an example, showing the multiple sequence alignment for the Furin-like domain, which is present in the Type 1 Insulin-like Growth Factor Receptor (1IGR) protein. Click anywhere on the image to open the complete, interactive CDD record for that domain model, cd00064. A separate section of this help document provides additional information about multiple sequence alignment display options.
In the CDD database, protein sequences from three-dimensional structures are included in domain models whenever possible, as one goal of the NCBI conserved domain curation effort is to make multiple sequence alignments agree with what we can infer from three-dimensional structure and three-dimensional structure superposition, in order to understand sequence/structure/function relationships.
The sequence-based domain models and corresponding 3D structures are also cross-referenced to each other through Entrez "Links" between CDD and structure records.
Conserved Domains can be described by local multiple sequence alignments (illustration) spanning a variety of organisms to reveal sequence regions that contain the same, or similar, patterns of amino acids. Computational biologists from all over the world have compiled collections of such alignments representing conserved domains. CDD includes domains curated at NCBI as well as data imported from the external sources listed below, and data sources are indicated by their accession number prefixes.
The source databases differ in their scope of coverage and the method by which they develop their models. Therefore, each source database may have its own model for a given conserved domain, in addition to some domain models found only in that database. To provide a non-redundant view of the data, CDD clusters similar domain models from various sources into superfamilies. The data sources include:
NCBI-Curated Domains
NCBI-curated domains use 3D-structure information to explicitly to define domain boundaries, aligned blocks, and amend alignment details. More details about the unique features of NCBI-curated domains are below.
The goal of the curation project is to provide CDD users with insights into how patterns of residue conservation and divergence in a family relate to functional properties, and to provide useful links to more detailed information that may help to understand those sequence/structure/function relationships. The presence of conserved features help to affirm family membership in search results with borderline significance, for example. NCBI CDD Curators provide feature annotation and associated evidence in a computer friendly way, so that the scientific community can build software tools for the automation of tasks like annotation transfer, for example.
NCBIfams
NCBIfams is a collection of protein family hidden Markov models (HMMs) for improving bacterial genome annotation. A paper by Haft et al. (2018) provides additional information about NCBIfams, which is part of NCBI's Reference Sequence (RefSeq) project.
External Data Sources
In addition, CDD imports data from five other major sources, below. The version number (as available) of each source database that is imported into CDD is provided in the CDD News page.
| Abbreviation |
Database Name |
Description |
| SMART |
Simple Modular Architecture Research Tool |
SMART is a web tool for the identification and annotation of protein domains, and provides a platform for the comparative study of complex domain architectures in genes and proteins. SMART is maintained by Chris Ponting, Peer Bork and colleagues, mainly at the EMBL Heidelberg. CDD contains a large fraction of the SMART collection. |
| Pfam |
Protein families |
Pfam is a large collection of multiple sequence alignments and hidden Markov models covering many common protein domains and families. Pfam is maintained by Alex Bateman and colleagues, mainly at the Wellcome Trust Sanger Institute. CDD contains a large fraction of the Pfam collection. |
| COGs |
Clusters of Orthologous Groups of proteins |
COGs is an NCBI-curated protein classification resource. Sequence alignments corresponding to COGs are created automatically from constituent sequences and have not been validated manually when imported into CDD. |
| TIGRFAMs |
The Institute for Genomic Research's database of protein families |
TIGRFAMs, a research project of the J. Craig Venter Institute, is a collection of manually curated protein families from The Institute for Genomic Research and consists of hidden Markov models (HMMs), multiple sequence alignments, Gene Ontology (GO) terminology, cross-references to related models in TIGRFAMs and other databases, and pointers to literature. |
| PRK |
PRotein K(c)lusters |
Protein Clusters is an NCBI collection of related protein sequences (clusters) consisting of Reference Sequence proteins encoded by complete prokaryotic and chloroplast plasmids and genomes. It includes both curated and non-curated (automatically generated) clusters. |
CDD also contains data from additional research projects, such as KOGs (a eukaryotic counterpart to COGs) and the Library of Ancient Domains (LOAD), contributed by I. Aravind, E. Koonin, and colleagues. The KOGs data set is accessible as a separate CD-Search database/Batch CD-Search database, and the LOAD data set is available on the FTP site, but neither of those data sets is directly searchable by text term in Entrez CDD.
The content of imported domain models is determined by the providers of the source database, with slight modifications made at NCBI to link a domain model's member sequences to corresponding, complete protein sequence and 3D structure records in Entrez databases, when possible. The method by which imported domain models are integrated into the CDD database is described in the CD assembly process section of this help document.
Accession Prefixes indicate data sources:
Source databases are evident from CD accessions:
| Accession starts with: |
Source Database |
| cd |
Curated at NCBI |
| sd |
Domain models specifically built to annotate structural motifs;
this is a subset of the NCBI-curated domain models. |
| NF |
NCBIfams |
| pfam |
Pfam |
| smart |
SMART |
| COG |
COGs |
| KOG |
KOGs (available as a separate search set via CD-Search (RPS-BLAST); not searchable by text term in Entrez) |
| PRK |
PRotein K(c)lusters (Entrez database) |
| CHL |
Chloroplast and organelle proteins; subset of the PRK database. |
| MTH |
Mitochondrial proteins; subset of the PRK database. |
| PHA |
Phage proteins; subset of the PRK database. |
| PLN |
Plant-specific (non-chloroplast) proteins; subset of the PRK database. |
| PTZ |
Protozoan proteins; subset of the PRK database. |
| TIGR |
TIGRFAMs |
| LOAD_ |
Library of Ancient Domains (LOAD) data set. (available as a separate data set via FTP; not searchable by text term in Entrez) |
Accessions that start with "cl" are for superfamily cluster records, which can contain domain models from one or more source databases.
When searching CDD, it is possible to limit search results to domains from any given source database by using the Database Search Field.
NCBI-curated domain models are assembled using the methods briefly described in the source databases section of this document. More details about the NCBI curation process are provided by Marchler-Bauer, et al. (2007). An example of a multiple sequence alignment on which a model is based is shown in an illustration of the Furin-like domain.
Domain models from external data sources are assembled by various methods, ranging from automated processing to manual curation, depending on the individual source database. Upon import into CDD, protein sequence alignments (illustration) from each of the source databases are processed in an automated way to provide links from each aligned sequence to the corresponding, complete record in the Entrez Protein database. Occasionally, sequences that cannot be identified in Entrez's databases are omitted or substituted for closely related matches. Whenever possible, sequences in PFAM, SMART, and COGs alignments are substituted for closely related sequences (passing a stringent sequence similarity threshold) that have direct links to three-dimensional structures in the Moleclular Modeling Database (MMDB).
A representative sequence is chosen for each domain model, preferably with a structure-link, for technical reasons. The representative sequence is generally shown as the first member of the multiple sequence alignment for a domain model. By default, this representative is the 3D structure shown when CD alignments are visualized with Cn3D.
A consensus sequence is computed from the imported alignments.
Alignment columns have to be represented in at least 50% of all aligned sequences (weighted by diversity) to determine the extent of the consensus. The most frequently occurring residue in each column (after weighting to account
for redundancy) is reported.
A position-specific scoring matrix (PSSM) is calculated for the extent of the consensus sequence. The PSSM profiles the various amino acids that were present in a given position of the multiple sequence alignment for a domain model and how frequently each one was observed. The consensus sequence does not contribute to the residue frequency statistics. Each PSSM receives a unique identifier (PSSM ID).
A PSSM ID is the unique identifier for a domain model's position-specific scoring matrix (PSSM). If a domain model's PSSM changes in any way as a result of updates to its multiple sequence alignment, it receives a new PSSM ID. This happens because a conserved domain model can evolve over time. For example, as new sequence data become available, the curators of a source database might add sequences to a multiple sequence alignment or update the sequences already present. As a result of such changes to the domain model, the PSSM and its ID can change. (Additional notes: Each superfamily record in the Conserved Domain Database also has a PSSM ID, which refers to the specific set of conserved domain PSSM IDs that comprise the superfamily, rather than to an actual position-specific scoring matrix for the overall superfamily. Obsolete PSSMs (e.g., 667) cannot be retrieved through the Entrez CDD search interface because they are no longer indexed. However, they can be retrieved from the archival copy of the database by using the "Direct Fetch via UID" option on the CDD Search Methods page.)
Search databases compiled of these PSSMs are available through the CD-Search
service (see help document) and on the NCBI FTP site as collections of pre-computed RPS-BLAST databases that can be used for locally installed versions of that program.
|
As noted in the section on CDD data sources, NCBI-curated domains use 3D-structure information to explicitly to define domain boundaries, aligned blocks, and amend alignment details.
The goal of the NCBI conserved domain curation project is to provide database users with insights into how patterns of residue conservation and divergence in a family relate to functional properties, and to provide useful links to more detailed information that may help to understand those sequence/structure/function relationships. To do this, CDD Curators include the following types of information in order to supplement and enrich the traditional multiple sequence alignments that form the foundation of domain models:
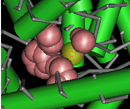
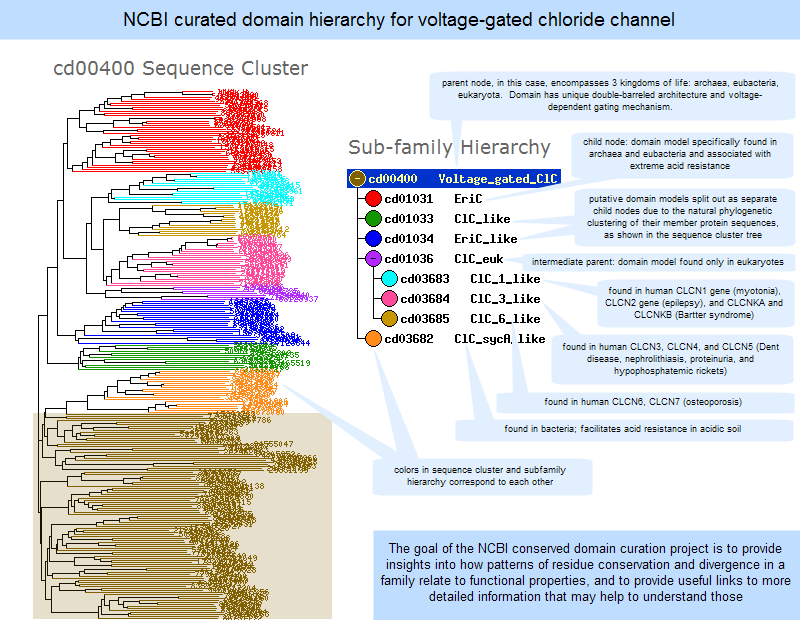
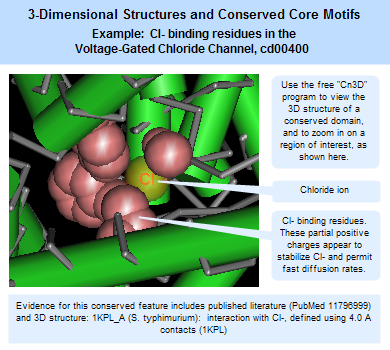
3-dimensional structures and conserved core motifs: NCBI Conserved Domain Curators have re-evaluated and modified multiple sequence alignments imported from outside sources, and made them agree with what we can infer from three-dimensional structure and three-dimensional structure superposition. Curated alignments contain aligned blocks spanning all rows (with no gaps allowed inside blocks) and unaligned regions between blocks. The blocks are meant to represent conserved structural core motifs of the corresponding domain family. The 3D structures can be viewed interactively with the Cn3D structure viewing program. More information about viewing structures is provided in the section of this document on CD summary pages, and the illustration at the right provides an example of a protein structure that has been annotated by NCBI curators to highight the Cl- binding residues.
(Click on the illustration to open the current, interactive record for the Voltage-Gated Chloride Channel domain model, cd00400, in the Conserved Domain Database (CDD). From there, you can open an interactive version of the 3D structure, with conserved feature annotations, in the free Cn3D structure viewing program.)
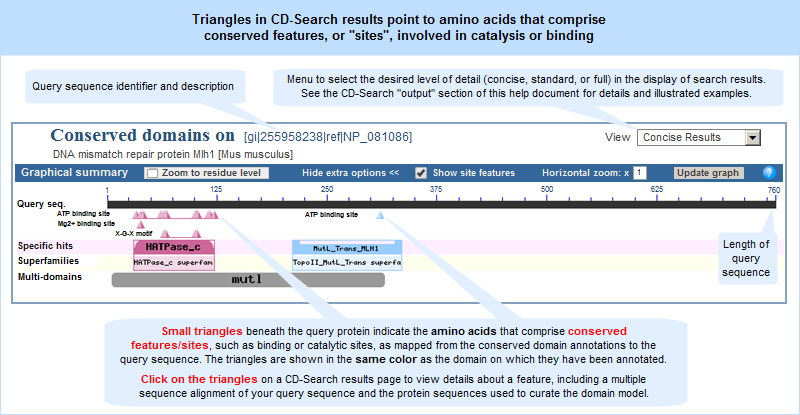
Conserved features/sites: In addition to working on the alignment model (illustration), NCBI curators also record, when possible, the location and nature of features conserved in the domain family. Typically these would describe catalytic residues, binding sites, or motifs commonly referred to in the literature.
|
|

Features are added if they seem applicable to the family described in the CD's scope and if there is evidence linking the feature to a set of addresses on the alignment. Such evidence is recorded and available for inspection; it may be free-text comments, citations linked to PubMed, or "structure evidence" - exemplifying the existence of a site by highlighting an actual molecular complex, for example. Both features and evidence can be visualized on CD summary pages (in the conserved features/sites summary box, and as hash marks (#) in the multiple sequence alignment displays), and with the Cn3D structure viewing program. An example is shown in the illustration at the right. (Click on the illustration to open the current, interactive record for the Voltage-Gated Chloride Channel domain model, cd00400, in the Conserved Domain Database (CDD). Note that the live web page may look different from the illustration shown here, because the Conserved Domain Database continues to evolve with the addition of new data; however, the concepts shown in the illustration remain stable.)
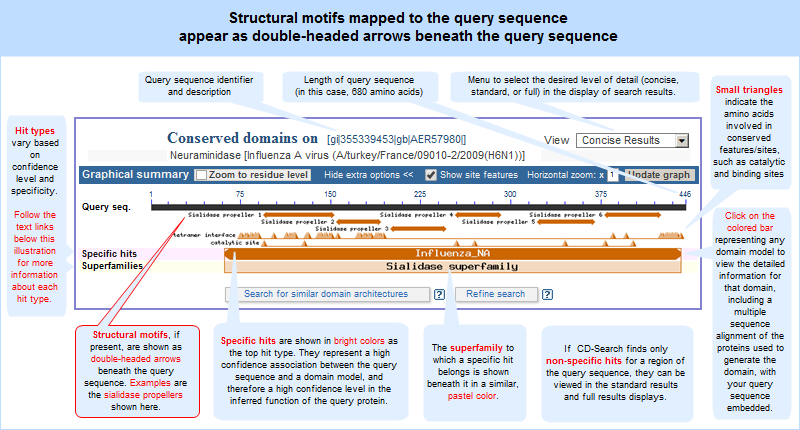
In addition, the CD-Search tool can be used to identify conserved features in a query protein sequence, designated by small triangles (illustrated example) in the search results graphical summary, when such features can be mapped from the conserved domain annotations to the query sequence.
Phylogenetic organization: Based on evidence from sequence comparison, NCBI Conserved Domain Curators attempt to organize related domain models into phylogenetic family hierarchies (illustrated example). The CDTree program used by NCBI curators can be downloaded in order to view NCBI-curated domains interactively and in greater detail.
Links to electronic literature resources: NCBI curated domains also provide links to citations in PubMed and NCBI Bookshelf that discuss the domain. These references are selected by curators and, whenever possible, include articles that provide evidence for the biological function of the domain and/or discuss the evolution and classification of a domain family.
NCBI-curated domains can be recognized in CDD search results by their "cd" accession number prefix. It is also possible to limit CDD search results to domain models from any given source database by using the Database Search Field.
|
A domain family hierarchy is a set of related domains that share a common ancestor, a common set of conserved residues, and a common general function, but differ from each other in their specific phylogeny, specific functions, and additional spans of conserved residues. Domain hierarchies are present in NCBI-curated domains in order to provide insights into how patterns of residue conservation and divergence in a family relate to functional properties.
Some domain families have only a single node, while others have a hierarchy that is two or more levels deep, sometimes with numerous nodes ("subfamilies")at each level. Such hierarchies have generic "parent" models and more specific "children". The parent node contains a span of conserved residues that is also present in each of the children. Each of the child nodes can have additional conserved residues that extend beyond that span and help to further characterize the members of the child node.
NCBI CDD Curators attempt to split "children" nodes where they see evidence for ancient gene duplications resulting in orthologous groups, often occurring together with functional divergence. The CDTree program used by NCBI curators can be downloaded in order to view NCBI-curated domains interactively and in greater detail, with or without a query sequence embedded.
An illustrated example of a subfamily hierarchy is provided below.
|
Click anywhere on the image to open the current, interactive record for the Voltage-Gated Chloride Channel domain model, cd00400, in the Conserved Domain Database (CDD). Note that the live web page may look different from the illustration shown here, because the Conserved Domain Database continues to evolve with the addition of new data; however, the concepts shown in the illustration remain stable.
|
A superfamily cluster is a set of conserved domain models that generate overlapping annotation on the same protein sequences. These models are assumed to represent evolutionarily related domains and may be redundant with each other. A superfamily accession number begins with the prefix "cl" for "cluster". (Some superfamilies contain only a single conserved domain model (singleton), and these are not indexed in Entrez. Only superfamilies that contain two or more conserved domain models are indexed in Entrez and will therefore appear in search results.)
Clustering methodology:
Superfamily members are clustered through an automated process that involves the following steps:
- Identify domain models that have overlapping hits on sequences in the Entrez Protein database from at least five different identical protein groups (IPGs).
Technical note: In the data processing pipeline at NCBI, protein sequence records that contain an identical sequence, regardless of TaxID, are placed in an identical protein group (IPG), and each group is given a stable unique identification number (referred to as IPG ID or UID).
- Store the overlapping domain models as pairwise associations, and use those pairwise associations to populate a similarity matrix.
- Refine the similarity matrix by comparing it against a "blacklist" (to remove unacceptable pairwise associations), and against a "whitelist" (to add pairwise associations that known to be valid but are not yet listed):
- The "blacklist" is used to separate domain models that should never be paired. The blacklist overrides all other aspects of the clustering algorithm.
For example:
- cd00538 (PA domain hierarchy) is blacklisted against pfam00082 (a serine protease model, subtilase family)
- cd00538 (PA domain hierarchy) is blacklisted against cd08022 (an M28-family metalloprotease)
These pairings above are forbidden because the PA (protease-associated) domain is often inserted in a protease domain, yet the proteases that contain the insert are distinct from each other. For example, if a PA domain is in a serine protease and also in a metalloprotease, the two types of protease would be clustered together by the algorithm in the absence of a black list. However, the metalloprotease and serine protease are actually distinct, and simply represent convergent evolution to similar function.
- The "whitelist" is used to add pairwise associations of domain models that are known to be related, but that might not have been listed in the initial, unrefined similarity matrix.
For example:
- NCBI-curated domain models that are organized hierarchically are part of the white list.
- Conserved domain models from external databases can also be grouped together, if those domains are known to be related but were not grouped automatically by the clustering algorithm. An example of such a whitelisted pair is shown in cluster cl23875: MvaI_BcnI Superfamily, which includes pfam15515 (MvaI/BcnI restriction endonuclease family) and pfam09562 (LlaMI restriction endonuclease), as of 30 January 2018. Those two conserved domain models for restriction endonucleases (REs) have relatively few hits in common. RE superfamilies are very sequence-diverse, meaning that models for specific subfamilies can differ quite a bit in terms of overall length and conserved residue patterns/signatures. Nevertheless, the models are whitelisted together because they are known to be related.
- Take the refined similarity matrix and feed it to the Python "fastcluster" package (https://pypi.python.org/pypi/fastcluster), to create clusters using the "complete linkage" algorithm.
- Implement a post-processing step to compare the Python-generated clusters against the whitelist, in case Python did not put two domain models in the same cluster but should have.
NOTE: Multi-domain models that were computationally detected are not included in Superfamily clusters. These models are likely to contain multiple single domains and might falsely join superfamily clusters.
Rationale:
Superfamilies provide a method for organizing data within CDD in a non-redundant way. CDD contains conserved domains from a number of different source databases, each of which may have its own model for a given conserved domain. The models might share many similiarities in their reported residue conservation patterns, but differ in the specific protein sequences used in the multiple alignment, their footprint length [domain boundaries], and biological annotations. Because of the similarities, RPS-BLAST might find that multiple domain models align to the same general region of a query protein, but have different footprints and E-value scores relative to the query protein. If the footprints of two or more domain models overlap on the query, those models are clustered into the same superfamily, then the superfamily continues to be extended using the methodology described above.
Example:
One example of a superfamily is Cluster ID cl02915, which contains various domain models for the voltage-gated chloride channel. Superfamily members include the NCBI-curated domain cd00400 and all members of that family hierarchy plus domain models from external resources.
Selection of Superfamily Representative:
A superfamily can contain one to many domain models. As of spring 2008, approximately 70% of the ~9,000 superfamilies contain a single model and the rest contain multiple models. Single model superfamilies often represent proteins specific to certain organisms or taxonomic lineages (for example, viruses). The numbers of superfamilies containing single or multiple domain models will continue to evolve as new domains are imported and new NCBI-curated hierarchies are added.
In superfamilies contatining multiple domain models, one of the models is selected as the source of the superfamily name and description. The representative is one of the following, listed in priority order:
- the parent node of an NCBI-curated domain family hierarchy, if one is present in the superfamily cluster. In the few cases where a superfamily contains more than one NCBI-curated domain, the parent of the hierarchy with the largest number of sequence hits is chosen as the superfamily representative.
- the Pfam domain model that hits the largest number of Entrez protein sequences in an RPS-BLAST search
- the SMART, COG, PRK, or CHL model that hits the largest number of Entrez protein sequences in an RPS-BLAST search
- the sole member of a superfamily
Superfamily can change over time:
The composition of a cluster can change over time due to a variety of factors, such as:
- availability of new domain models as the Conserved Domain Database continues to grow
- changes to previously existing models
- new and/or updated sequence records in the Entrez Protein database
- refinements to the automated clustering procedures
A superfamily cluster accession number will remain the same if at least 50 percent of its member models (conserved domain accessions) have not changed relative to the previous version of the cluster.
If more than 50 percent of the conserved domain accessions from a previous version of a cluster are no longer present in the new build of that cluster, or if the cluster size more than doubles with a new build, then the superfamily cluster accession is retired and replaced by a new accession(s). If two previous clusters merge into a single new cluster, the superfamily cluster accession number of the larger component cluster is used for the new grouping.
A superfamily also has a PSSM ID, which refers to the specific set of PSSM IDs for the domain models that comprise the superfamily, rather than to an actual position-specific scoring matrix for the overall superfamily. The superfamily PSSM ID will change if there is any change to the set of member PSSM IDs relative to the previous version of the cluster (e.g., if a member conserved domain gets a new PSSM ID due to changes in its multiple sequence alignment, of if a new conserved domain model is added to the superfamily as the result of a CDD database update).
The CD summary page for a Superfamily record does not include a multiple sequence alignment display; rather, it provides the name and description of the superfamily and lists the domain models that belong to it. The multiple sequence alignment for any member domain model can be viewed by clicking on it to open its CD summary page.
Superfamilies that contain a single domain model ("singletons")
The concept of superfamilies was applied to CDD in order to cluster related conserved domain models together and provide a non-redundant view of the available domain models. After the superfamily clustering algorithm is applied to the domain models in CDD, all resulting clusters are referred to as superfamilies, regardless of how many domain models they contain. The non-redundant view of CDD therefore includes superfamilies with a single domain model ("singletons") as well as superfamilies containing two or more domain models.
In the user interface, however, superfamilies that contain only one model are not displayed in search results, or listed as links from the domain model, because they look very similar to the model itself.
In contrast, superfamilies that contain two or more models ("multi-model superfamilies") are displayed in search results, and are also accessible as links from their member domain models. The number of multi-model superfamilies is provided in the Database Statistics box on the "Conserved Domains and Protein Classification News" page, and they can be retrieved by clicking on that statistic.
| Protein query sequence | Text term search in Entrez CDD | Protein → Conserved Domains links | Domain architecture |
Protein Query Sequence (CD-Search):
Most users will explore conserved domains starting from CD-Search results for a protein of interest.
The query can be a protein sequence in FASTA format or the GI or Accession of a protein sequence that exists in the Entrez Protein database.
The search results will show the conserved domains found in the protein. The colored bars that depict the domain footprints (shown in both the concise display and full display of CD-Search results) are active hotlinks that open the corresponding CD summary pages with your query sequence embedded in the multiple sequence alignment of proteins used to create the domain model.
The second half of this help document provides details on how to use the CD-Search service, including input required and output shown.
Text Term Search in Entrez CDD:
| allowable search terms | search methods: basic and advanced | search fields | quotes | wild card * |
| search results | document summary page | "display", "show, "sort by", "send to" menus) |
Allowable search terms
Conserved domains can be searched by text term in the
Entrez CDD database. The Entrez query interface allows searching for keywords, publication dates, and taxonomic span, accesssion numbers, and more. The search field summary table in this document shows the variety of terms that can be used to query the database and provides sample searches. It is also possible to use quotes to force multiple terms to be searched as a phrase, and to use an asterisk (*) as a wild card to search for a word stem.
For example, search the Entrez CDD database for strings like "Kinase" or "pfam023*" or "Tetratrico*" to see how it works:
|
A number of techniques can be used to search the database, offering varying degrees of control over your query. The search methods summary table provides examples of basic and advanced searches. In basic searches, you can just enter one or more search terms without specifying search fields, Boolean operators, or other search criteria. These searches are quick and easy but can result in some extraneous hits. Advanced search methods, on the other hand, allow you to exercise greater control over your search, for example, by specifying which search field to use for each query term, limiting search results to a particular type of record or source database, or refining your search in other ways. A separate section of this help document describes the CDD search results.
(The PubMed help document and Entrez help document provide additional, general information about using the Entrez search system.)
Search Methods
A variety of techniques can be used to search the Entrez CDD database, offering varying degrees of control over your query. In some cases, they offer alternative ways of executing the same search (as is true for sample searches #4, #5, and #6 below), with each method offering different benefits. The search methods include:
| |
| Method |
Description |
Example |
| Basic Search |
Just enter search terms without specifying search fields, other limits, or Boolean operators.
The "Search Details" box in the right margin of the search results page shows exactly how Entrez parsed and handled your query. If desired, you can edit the query in that box and press the "Search" button to run the modified query.
The "See more..." link a the bottom of the "Search Details" box opens a more detailed display:
- The Query Translation box shows the search strategy used to run the search
- To edit the search in the Query Translation box, add or delete terms and then click Search.
- Click URL to display the current search as a URL to bookmark for future use. Searches created using History numbers can not be saved using the URL feature.
- You may also save your search using My NCBI.
- The Result number link retrieves the documents found and displays them in a search results page.
- Translations details how each term was translated using Entrez's search rules and syntax for the database.
- User Query shows the search terms as you entered them in the search box and any syntax errors with the query.
|
Search #1:
mismatch repair eukaryotes
will retrieve biosystems with those terms anywhere in the record.
Some of the records might include aligned sequences from organisms other than eukaryotes because we did not limit that search term to the Organism search field. Because of this, we might also retrieve conserved domain records they happen to contain the term "eukaryotes" in a comment or some other field of the record.
Similarly, the term "mismatch repair" can appear anywhere in the record.
The terms entered in a basic search may or may not be adjacent to each other in the retrieved records, depending on how Entrez parsed the query (as shown in the Search Details for a given search). To force terms to be searched as a phrase, use quotes. To refine your search in other ways, use the Limits option or the Advanced Search methods described below.
|
Limits |
The Limits page allows you to restrict your search in various ways.
At a minimum, the Limits page displays the list of available search fields. You can do a separate search for each term or phrase in your query, as shown in sample Search #2 and #3 to the right, and select the desired search field for each one. (If desired, you can then combine the searches by using the Search Builder or History section of the Advanced Search page.)
For some databases, the Limits page also provides other commonly used options, as check boxes and/or pull-down menus, for restricting your search results to records with specific characteristics. These check boxes and pull-down menus generally represent a commonly used subset of the choices that are available from the Advanced Search page and are placed on the Limits page for easy access.
IMPORTANT NOTE: Once you have used a particular Limit, warning sign will appear near the top of your search results page that indicates which Limit(s) are currently in effect, for example:

Note that the Limit will remain in effect for all subsequent searches in the current database unless you change or remove that limit. In the illustrated example above, any search you do will be limited to the Titles of records, until you remove the limit.
|
Search #2:
On the Entrez CDD search page, click on the Limits link, select the Text Word, enter the following query:
"mismatch repair"
and press "GO". That will retrieve only records which contain those terms in the conserved domain's description. The quotes surrounding the terms force them to be searched as a phrase.
Search #3:
Open the Limits page again and clear your previous search. Change the search field selection to Organism,
enter the following query:
eukaryotes
and press "GO". That will retrieve conserved domain models containing only eukaryotic sequences in their multiple sequence alignments (i.e., eukaryota will be the root taxonomic node of the sequences in a domain model's alignment).
If desired, you can then combine the searches on the Advanced Search page, either by using the Search Builder, as shown in sample Search #4, or by using the History section of that page, as shown in sample Search #5.
|
| Advanced Search |
The Advanced Search page allows you to exercise greater control over your search, for example, by enabling you to:
- Build a search one step at a time.
- Browse the index of any search field and add term(s) of interest from the index to the active query box at the top of the page.
- View your search History and combine or subtract searches from each other.
As you build a query, either by using the Search Builder's pull-down menus, or by using the "Add" links in the "History" portion of the page to combine previous searches, the grey text box at the top of the page will display your current query.
You can also manually edit the current query by clicking the "Edit" link beneath the grey text box. That will allow you to type terms/search numbers/etc. directly into the box, add parentheses for nesting if desired, change Boolean operators, etc.
In addition, the following types of advanced searches can be entered in the query box of any Entrez search page (i.e., in the query box of the database's Home page, Limits page, or Advanced Search page):
|
Search Builder |
The "Search Builder" section of the Advanced Search page allows you to build your query step by step, adding a new search term and selecting a new search field at each step. It also allows you to browse the index of any search field to view the available terms.
To build a query:
(1) Select the Search Field of interest using the pull-down menu.
(2) Type a term(s) in the text box beside the search field menu. Or, use the "Show index list" link to see the index of the search field and select the desired term from the index. (tips on using the "Show Index List")
(3) Select the Boolean operator (AND, NOT, OR) that should precede the term when it is added to the active query at the top of the page.
Continue the above steps, as desired, to add more term/search field combinations to your query.
As you use the Search Builder, the grey text box at the top of the page will show your current query.
You can manually edit the current query by clicking the "Edit" link beneath the grey text box. That will allow you to type terms/search numbers/etc. directly into the box, add parentheses for nesting if desired, change Boolean operators, etc.
Press the Search button to display the records retrieved by your search (i.e., it displays the search results page).
Click on the "Add to history" link if you prefer to simply add the query to your search history and remain on the Advanced Search page, where you can continue building your query.
Tips on using the "Show Index List" function on the Advanced Search page:
The "Show Index List" function allows you to browse the index of any Search Field. If you select a search field and press the "Show Index" link without entering a term in the box, you will be taken to the top of the index. If you enter a term first, you will be taken to the part of the index that contains your term (or the closest alphabetical location, if your term is not present in the index).
The number of records that contain the term will appear in parentheses. You can also browse the index to explore the variety of terms available (for example, select "All Fields", enter "Huntington", and click on the "Show Index" link to see additional spellings and/or related terms, such as Huntington disease, Huntington's, Huntington's disease).
To select a range of terms from the index, use the Shift key while selecting the first and last term. Then use the AND, OR, or NOT buttons to add that group of terms to the active query.
To select multiple terms that do not fall within a continuous range from the index, use the Control key while selecting the terms of interest. Then use the AND, OR, or NOT buttons to add that group of terms to the active query.
Note: When multiple terms are selected from the index window, they are OR'ed together within parentheses and then appended to your query with whatever Boolean operator you have selected.
|
Search #4:
On the Entrez CDD search page, click on Advanced Search and build your search one step at a time:
(a) Using the first pull-down menu in Search Builder, select the Text Word search field and enter the following query:
"mismatch repair"
and select "AND" as the Boolean operator. That term/search field selection will automatically be displayed in the grey text box at the top of the page, which shows your current query.
(b) Using the second pull-down menu in Search Builder, select the Organism search field and enter the following query:
eukaryotes
and select "AND" as the Boolean operator. That newest term/search field selection will automatically be added to the grey text box at the top of the page.
(c) Your query will now appear as:
"mismatch repair"[Text Word] AND eukaryotes[Organism]
Press the Search button if you want to display the records retrieved by your search (i.e., it displays the search results page).
Or, click on the "Add to history" link if you prefer to just add the query to your search history and remain on the Advanced Search page, where you can continue building your query.
Note that this search will produce the same results as sample searches #5 and #6. It is simply executed in a different way. That is, you remain on a single query page (Advanced search) and can browse the index of any search field as you build your query one step at a time.
|
History |
The "History" section of the Advanced Search page displays the searches you have done in the current database.
You can combine or subtract searches from each other by entering the search numbers and the AND, OR, or NOT Boolean operators in the query box, for example: #2 AND #3. If the query contains several search numbers and Boolean operators, the Boolean operators are processed from left to right unless parentheses are used for nesting. If parentheses are used, the portions of the query in parentheses will be processed first, then the remaining Boolean operators will be processed from left to right.
Additional details about Search History:
- The Search History will be lost after 8 hours of inactivity. (To save a search indefinitely, click on the search # and select "Save in My NCBI.")
- Click "Clear History" to delete all searches from History.
- Entrez will move a search statement number to the top of the History if a new search is the same as a previous search.
- History search numbers may not be continuous because some numbers are assigned to intermediate processes, such as displaying a citation in another format.
- The maximum number of searches held in History is 100. Once the maximum number is reached, PubMed will remove the oldest search from the History to add the most current search.
- A separate Search History will be kept for each database, although the search statement numbers will be assigned sequentially for all databases.
- PubMed uses cookies to keep a history of your searches. For you to use this feature, your Web browser must be set to accept cookies.
- Database records that you have copied to the Clipboard are represented by the search number #0, which may be used in Boolean search statements. For example, to limit the records you have collected in the Clipboard to those from human, use the following search: #0 AND human[organism]. This does not change or replace the Clipboard contents.
|
Search #5:
Use the search numbers shown in the "History section" of the advanced search page to combine previous searches (for example, searches #2 and #3 shown above).
To do that, you can either:
Click on the "Edit" link beneath the grey text box and type in a search statement such as:
#2 AND #3
Or, instead of typing the search statement, use the "Add" link beside any search number in the "History" section of the Advanced Search page to add that search number into the grey text box.
That will retrieve records containing "mismatch repair" in the Text Word field and "eukaryotes" in the Organism field. Compare the retrieval from this search with that of the sample basic search above.
(Note that your search numbers might be different from those shown here, if you did earlier searches in the Entrez system before trying these examples.)
|
| Complex Boolean |
Whether you are on the Basic search page (i.e., the database's home page), the Limits page, or the Advanced search page, you can:
Enter a search in command language, specifying your exact combination of desired search terms, search fields, and Boolean operators, as shown in the examples to the right. The syntax is:
term[field] BOOLEAN term[field] BOOLEAN term[field] etc.
Search Field names must be placed in square brackets [], and can be written as either the full name, for example, [Database], or as the corresponding search field abbreviation, for example, [db] (additional examples).
Boolean operators (AND, OR, NOT) must be written in UPPER CASE.
Boolean operators are processed from left to right unless parentheses are used for nesting. If parentheses are used, the portions of the query in parentheses will be processed first, then the remaining Boolean operators will be processed from left to right.
Boolean operators can also be used to combine or subtract searches from each other (i.e., to find the union, difference, or intersection of the data sets retrieved by various searches). To do this, use the History section of the Advanced Search page and simply enter the search numbers and desired Boolean operators in the query box.
For example, to identify the records that were retrieved by Search #2 of your search history, and also by Search #3, you could enter the following query:
#2 AND #3
To identify the records that were retrieved by Search #2 but not by Search #3, you could enter the following query:
#2 NOT #3
|
Search #6:
Simply enter all search terms and search fields as a single statement into the query box:
"mismatch repair"[Text Word] AND eukaryotes[Organism]
Note that this search will produce the same results as sample searches #4 and #5, but it takes only a single step when entered directly into the search box as a Boolean query.
Search #7:
("chloride channel"[All] OR ClC[All]) AND (cdd[Database] OR pfam[Database])
This search will retrieve biosystem records that contain the phrase "chloride channel" or the abbreviation "ClC" in any field of the record, and that are from the NCBI-curated or PFAM source databases.
|
| Batch query |
If you have a list of conserved domain accession numbers or PSSM IDs, you can use Batch Entrez to retrieve them.
Be sure to select the appropriate database from the pull-down menu at the top of the page, then "Browse" to find the file of UIDs you'd like to upload, and press "Retrieve." Batch Entrez will then display a report summarizing the data it found in your input file, such as: (1) number of lines that were present in the file (there should be one UID per line); (2) rejected lines (indicating how many invalid UIDs were detected); (3) removed duplicates (indicating how many duplicate UIDs were detected/removed); (4) passed to Entrez (indicating how many of the UIDs from your list are valid and will therefore be acted upon). The latter may include obsolete UIDs that have been superceded by newer UIDs. The obsolete UIDs, as well as any invalid UIDs that were present in your file, are explicitly reported at the top of the Batch Entrez report.
If you have a list of protein sequences in which you'd like to identify conserved domains, you can use the Batch CD-Search service. See the Batch CD-Search help for additional information. (To search for conserved domains on a single protein sequence, the original CD-Search service continues to be available, along with the CD-Search help.)
|
|
Additional details about search methods and options are provided in the: (1) PubMed help document (including information about temporarily saving records from your search results to the Clipboard); (2) My NCBI help document (including information about Saving search strategies and indefinitely saving records from your search results into your My NCBI Collections); and (3) general Entrez help document.
|
|
Search Fields
By default, the Entrez system searches all fields of a record. If you want to narrow your query by searching for your term(s) in a specific search field, you can select the desired field by using the pull-down menus on either the Limits and Advanced search page, or you can type the search field directly in your query (surrounding field names with square brackets [], for example, [Organism] or [Orgn]).* The Show index link on the Advanced search page allows you to browse the index of each search field, where you can see the available terms, the number of records containing each term or phrase, as well as the syntax for entering values in search fields such as Modification Date or Publication Date.
The currently available fields include:
| |
| Field name |
Abbreviation* |
Description |
Sample Search |
| All Fields |
[all] |
Searches the complete database record |
"chloride channel"[All]
will retrieve the CDD records that contain the phrase "chloride channel" in any field of the record.
The quotes surrounding the search terms ensure they are searched as a phrase.** |
| Accession |
[accn] |
Searches only the accession number of the record, which is always an alphanumeric combination. The accession number prefix indicates the source database. The accession number applies to the complete conserved domain record.
Note: An additional unique identifier, the PSSM ID, is assigned to the position specific scoring matrix that is derived from the conserved domain's multiple sequence alignment. Conserved domains can also be retrieved by entering their PSSM ID (without a search field specifier). |
cd00400[Accn]
will retrieve the CDD record that contains the specified unique identifier in the accession number field. |
| Alternative Accession |
[AltAccn] |
Native accession format from an external source database. For example, the PFAM database uses accessions with a format such as pf08617. When these are imported into CDD, the accessions are represented in a format such as pfam08617. Similarly, the SMART database uses a format such as sm00100, while records that have been imported into CDD have a format such as smart00100. This is primarily done to indicate that SMART and PFAM domain alignments may have been modified slightly by NCBI staff, for example by the substitution of a protein sequence that does not have 3D structure with a highly similar one that does (as explained in the help document section on the CD assembly process). |
pf08617[AltAccn]
will retrieve the pfam08617 record from CDD. |
| Database |
[db] |
Use this field to limit your search to a particular source database. |
cdd[db]
will retrieve the NCBI curated domain models and superfamily records, which are also created at NCBI, from CDD.
pfam[db]
will retrieve the domain models that were imported from the PFAM database.
|
| Filter |
[filt] |
The "Filter" search field allows you to narrow your retrieval to records that have certain attributes, such as curated or uncurated, or records that have links to other Entrez databases of interest.
Many attributes from the Filter field are provided in the "Links" menus that are present on an Entrez search results page, and in the "Links" box on an individual CD Summary page. A detailed explanation of each type of link is provided in the description of the "Links" box. |
cdd_gene[filt]
will retrieve the CDD records that have associated data in the Entrez Gene database.
On the CDD search results page, you can then open "Display" menu and select the Gene Links option to view the corresponding Entrez Gene records. |
| Modification Date |
[mdat] |
Date of the most recent changes to the alignment model and/or descriptive information |
|
| Number of Sites |
[ns] |
The number of conserved features, such as catalytic or binding sites, that have been annotated on a domain. Conserved features are available on NCBI-curated domains.
As of April 2008, this ranges from zero to 21 sites. (To see the current range, select the "Number of Sites" search field on the "Search Builder" section of the "Advanced search" page, then use the "Show index" link to view the index of that search field and see available values.) |
4[ns]
will retrieve the NCBI curated domain models that contain four sites (i.e., four conserved features). |
| Organism |
[Orgn] |
The root taxonomy node of a conserved domain. This is the highest node in the NCBI Taxonomy database that encompasses all organisms whose protein sequences are in the multiple sequence alignment for a domain model. |
eukaryotes[orgn]
will retrieve conserved domains found in eukaryotes. |
| PSSM Length |
[plen] |
Length of the PSSM or domain search model. This is the same as the length of the consensus sequence. |
|
| Publication Date |
[pdat] |
the date on which a CD was published |
|
| Structure Representative |
[strp] |
The number of structures that have protein sequences in the multiple sequence alignment for a domain model.
As of January 2010, this ranges from zero to 70 protein sequences from structures. (To see the current range, select the "Structure Representative" search field on the "Search Builder" section of the "Advanced search" page, then use the "Show index" link to view the index of that search field and see available values.)
NCBI-curated domain models tend to have more structure representatives because the curation process includes incorporation of protein sequences from resolved structures. However, domain models from external data sources may also contain structure representatives. As noted in the section on data processing, sequences in PFAM, SMART, and COGs alignments are substituted, whenever possible, for closely related sequences that have direct links to three-dimensional structures in the Moleclular Modeling Database (MMDB). |
6[strp]
will retrieve domain models that contain six protein sequences from 3D structures in their multiple sequence alignment. |
| Subtitle |
[subtitle] |
The subtitle of a conserved domain, which may contain descriptive terms not present in the conserved domain's title.
An example of a title:subtitle combination is present in pfam00654:
"Voltage_CLC: Voltage gated chloride channel"
A search of the subtitle field for "chloride channel" will retrieve that record and others.
Note: Not all conserved domain models have subtitles, and search of the Text Word field may therefore retrieve more comprehensive results. |
"chloride channel"[subtitle]
will retrieve the CDD records that have the phrase "chloride channel" as part of their subtitle.
The quotes surrounding the search terms ensure they are searched as a phrase.** |
| Text Word |
[word] |
The long description (text summary) of the conserved domain. |
|
| The Description of Sites |
[sd] |
Brief descriptions of conserved features. |
|
| Title |
[titl] |
The short name of a conserved domain, which concisely defines the domain.
Example: "Voltage gated ClC" is the short title of the NCBI-curated conserved domain model (cd00400) for the voltage gated chloride channel.
Note: A search of this field will also retrieve superfamily clusters (cl* accessions) that contain one or more domain models with the search term in their short title, even if the title of the superfamily cluster itself does not contain the search string. |
voltage*[titl]
will retrieve the CDD records that have the term "voltage" as part of their short name, such as cd00400: Voltage gated ClC and pfam00654: Voltage CLC, which represent NCBI-curated and externally imported domain models, respectively, for the voltage gated chloride channel. (The asterisk (*) is a wild card that can be used to search for a word stem.) |
| UID |
[UID] |
Retrieves a conserved domain record by its PSSM ID. If you enter a string of digits as a query and do not specify a search field, the UID field will be searched by default.
Note: As mentioned in the section on CD Assembly Process, when domain models evolve as new sequence data becomes available, their PSSMs can change, and in such cases, they received new PSSM IDs. Obsolete PSSMs (e.g., 667) cannot be retrieved through the Entrez CDD search interface, even with direct searches of the UID field, because they are no longer indexed. However, those obsolete PSSMs can be retrieved from the archival copy of the database by using the "Direct Fetch via UID" option on the CDD Search Methods page. |
238233[UID]
will retrieve the conserved domain record cd00400, whose PSSM ID is 79359.
79359
will also retrieve that same conserved domain record, because the UID field is searched by default for queries that are only a string of digits.
|
* In a query, the field name may be typed as the full name or abbreviation, and may be in upper, lower, or mixed case. It must be surrounded by square brackets []. A space between the search term and the field specifier is optional. If desired, surround a phrase with quotes to force an adjacency search. For example, the sample queries below will work equally:
"chloride channel" [WORD]
"chloride channel"[WORD]
"chloride channel" [word]
"chloride channel"[Text Word]
** The quotes surrounding the search terms in the All Fields example ensure the terms are searched as a phrase. If quotes are not used and the terms are not automatically recognized as a phrase by the Entrez system, Entrez will insert a Boolean AND between the terms and they may or may not appear adjacent to each other in the retrieved records. More search tips are provided in the PubMed help document and Entrez help document.
It is also possible to search for a word stem by using an asterisk (*) as a wild card; for example, chlori* will retrieve records with terms such as chloride, chlorin, chlorinate, chlorite. The Entrez Help document provides additional information about truncating search terms in this way.
|
|
Search Results
| document summary page | display settings: format, items per page, sort by | send to | filter your results | find related data |
Document Summary (DocSum) Page
After querying Entrez CDD by text term, the initial search results page (also referred to as the document summary, or "DocSum") provides a list of the conserved domain records that contain your search terms. The terms can appear in any field of the record, unless a search field was specified in the query. (Note: A separate part of this document describes the results of a search by protein query sequence using CD-Search.)
Click on the accession number or thumbnail image of any record on the DocSum page to view its conserved domain (CD) summary page.
If desired, you can narrow your search by restricting the query to a search field of interest or adding more terms with a Boolean AND.
Alternatively, you can broaden your search by adding more terms (e.g., synonyms) to your query with a Boolean OR, or by following links to Superfamily Members.
| |
| Click on the image above to open the live search results page in CDD. Note that the number of items retrieved may be different than shown here because the Conserved Domain Database continues to evolve with the addition of new data. |
|
|
|
|
Display Settings:
The "Display settings" menu on acts upon all of the conserved domain records (default) in your search results, or on the subset you have selected with checkboxes. You can select items from multiple pages of the search results, if desired.
| Format |
Summary -- a summary of all of the structure records (default) retrieved by your search, or for those you have selected with checkboxes, in HTML format.The information shown for each record may include the following, as available:
- Short name, which concisely defines the conserved domain
- Thumbnail image indicating if the conserved domain includes a protein sequence from a 3D structure.
If a 3D structure is included, the thumbnail will be a still graphic of the actual domain structure.
If no 3D structure is available for the protein family from which the domain model was created, the thumbnail icon will show a schematic of a multiple sequence alignment.
- First 100 characters of the text summary, which provides a synopsis of biological function and salient features of the domain
- Accession number
- PSSMid
- A subset of links to additional information about the domain, including a "View in Cn3D" link that opens an interactive view of the domain's 3D structure in NCBI's free Cn3D structure viewing program and links to related data in other Entrez databases. (Note: The "Find Related Data" menu in the right margin of the search results page provides a complete list of links. That menu retrieves related data for all records (default) retrieved by your search, or for the subset of records you have selected with checkboxes.)
Summary (text) -- a summary of the records retrieved by your search, in plain text format. By default, all records from your search result are listed. If you are interested only in specific records, select their checkboxes, select the desired display settings, and press "Apply" to view only those records. The information shown for each record is the same as in the "Summary" format described above, but does not include the subset of links to additional information.
UI List -- a list of the unique identifiers (UI's) for all of the conserved domain records (default) retrieved by your search, or for those you have selected with checkboxes.
|
| Items per page |
By default, 20 documents are listed per page. If desired, decrease (to a minimum of 5) or increase (to a maximum of 200) the number of documents displayed per page then press the "Apply" button.
|
| Sort by |
Search results are displayed in order of decreasing relevance with respect to the query. Many search fields have a score or rank associated with them; for example, the Title and Organism fields have a high rank, while the Description of Sites field has a lower rank. The presence of a search term in any one or more of the fields is scored accordingly by the search system, and the total score given to a hit is used in determining its relevance to the query and therefore its placement on the search results page.
Additional options are available to sort records by descending or ascending order of Accession,
Database,
Modification Date,
Number of Sites,
PSSM Length,
Publication Date, and
Structure Representatives.
A few of these sort options will cause certain types of records to cluster at the top or bottom of the search results, depending on whether ascending (up) or descending (down) order is chosen. For example, if you sort by:
Number of Sites - NCBI-curated domain models will appear at the top of search results (if "sort by number of sites (down)" order is selected) because conserved features, such as catalytic or binding sites, are annotated only on those domain models.
PSSM Length - the superfamily records will appear at the bottom (if "sort by PSSM length (down)" is selected) because they do not have an actual Position Specific Scoring Matrix (PSSM). Rather, each member of a superfamily has a PSSM and corresponding PSSM ID. A superfamily's PSSM ID refers to the specific set of conserved domain PSSM IDs that comprise the superfamily, rather than to an actual position-specific scoring matrix for the overall superfamily.
Structure Representatives - NCBI-curated domain models will tend to appear at the top of search results (if "sort by structure representatives (down)" order is selected) because the curation process includes incorporation of protein sequences from resolved structures. Domain models from external data sources may also contain structure representatives. As noted in the section on data processing, sequences in PFAM, SMART, and COGs alignments are substituted, whenever possible, for closely related sequences that have direct links to three-dimensional structures in the Moleclular Modeling Database (MMDB).
Technical note: If you retrieve all records in the database by searching the Filter field for All[Filt], the records are simply displayed in descending order of UID (i.e., PSSM ID).
|
"Send To" menu options
The "Send To" menu options act upon all the hits retrieved by your search (default), or those you have selected by using their checkboxes.
| File |
Saves all the hits retrieved by your search into a plain text file, in either "Summary (text)" or "UI List" format.
|
| Clipboard |
Copies all the hits retrieved by your search (default), or those you have selected with check boxes, into a Clipboard, which temporarily stores up to 500 items (they will be lost after 8 hours of inactivity).
Click on the "Clipboard: XX items" link in the upper right corner of the page to view the items in any format for up to 8 hours after your last activity in the database.
The Clipboard will not add an item that is currently in the Clipboard; it will not create duplicate entries. You can remove items from the Clipboard, if desired.
Entrez uses cookies to add your selections to the Clipboard. For you to use this feature, your Web browser must be set to accept cookies.
Items in the Clipboard are represented by the search number #0, which may be used in Boolean search statements. For example, to limit the items you have collected in the Clipboard to those from human, use the following search: #0 AND human[organism]. This does not affect or replace the Clipboard contents.
The Clipboard's "Send to" menu offers you the same "File" and "Collections" options as offered on the original search results page. The latter option saves all items (default), or the subset of items selected with check boxes, indefinitely in the My NCBI Collections section of your My NCBI account.
|
| Collections |
Saves all the hits retrieved by your search (default), or those you have selected by using their checkboxes, into the My NCBI Collections section of your My NCBI account.
|
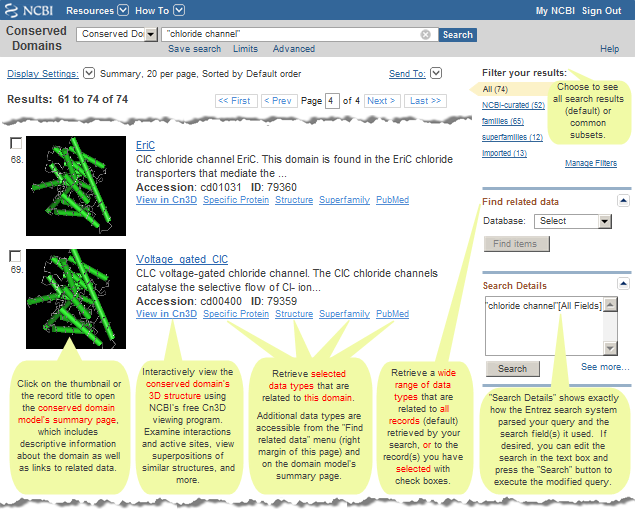
Filter your results
The "Filter your results" area in the upper right corner of a search results page allows you to see all the records (default) retrieved by your search, or subsets of your search results that reflect commonly requested categories of records, and shows the corresponding number of records in each case.
The links for "NCBI-curated," "imported," "families" (individual conserved domain models), and "superfamilies" (clusters of evolutionarily related conserved domain models into which the individual conserved domain models fall) show the number of retrieved records that fall into each of those categories, and allow you to view those subsets of your search results, if desired.
Find related data:
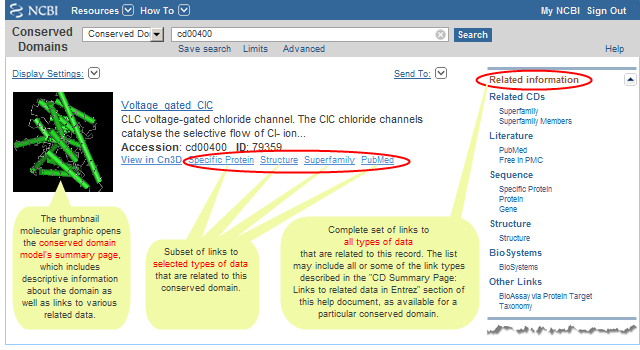
The "Related information" box that appears in the right margin of the display for an individual record allows you to retrieve related data for that particular domain model. (For example, the "Related CDs/Superfamily Members" link for accession cd00400 will retrieve the other domain models in the Conserved Domain Database that appear to be evolutionarily related to or redundant with cd00400.)
A "Find Related Data" box (instead of an "Related information" box) will appear in the right margin of a CDD search results page if you retrieved two or more records. The "Find Related Data" box allows you to retrieve related data for all the models retrieved by your search (default), or for the domain models you have selected with checkboxes. (For example, the "Find Related Data" option for Related CDs/Superfamily Member Links will retrieve the other domain models in CDD that appear to be evolutionarily related to or redundant with the domains retrieved by your search, or with the domains you have selected with checkboxes.)
The links in either display can include the following, depending on the related data that are available for the domains you have retrieved:
A "Links" box also appears in the displays of individual conserved domain records. All links are described in the help document section on "CDD Record (CD summary page): What information is displayed for each domain model on its CD Summary page?" : "Links to related data in Entrez". The number and type of links that exist vary among CDD records, depending on the related data that are available for any given record.
Most links are accessible on both the search results page and on a CD summary page, although a few of the links are available in only one of those places (*), such as Representatives and Books links, which are available only on the CD summary page.
Entrez Protein links to Conserved Domains:
Another (indirect) way to search the Conserved Domain Database is to start in a database such as Protein or Structure and use the "Find Related Data" ad in the right margin of the search results page, or use the "Related Information" ad in the right margin of the display for an individual record, to traverse to conserved domains.
For example, all sequence records in the Entrez Protein database have been RPS-BLASTed against the Conserved Domain database. These pre-calculated search results are available as "Conserved Domains" links from protein sequence records, making protein functional information one click away from the sequence record.
Several different types of links are available, allowing you to choose:
(a) the format in which you want to view the conserved domains (e.g., in graphical format as domain footprints aligned to the protein sequence, or as a list of records from the Conserved Domain Database, each of which includes a multiple sequence alignment of the proteins used to create the domain model), and
(b) the level of redundancy in the list of conserved domain models (e.g., a concise list of the top scoring models or a full list of all models that have a statistically significant RPS-BLAST hit to the protein).
The number of conserved domain models retrieved, and the order in which they are sorted/presented, depends upon the view you select:
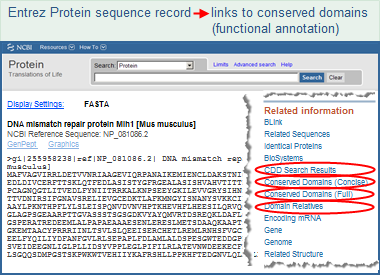
Click on the image to open the actual Entrez protein sequence
record and follow the live links to conserved domains.
You may need to scroll down the display to see the links.
|
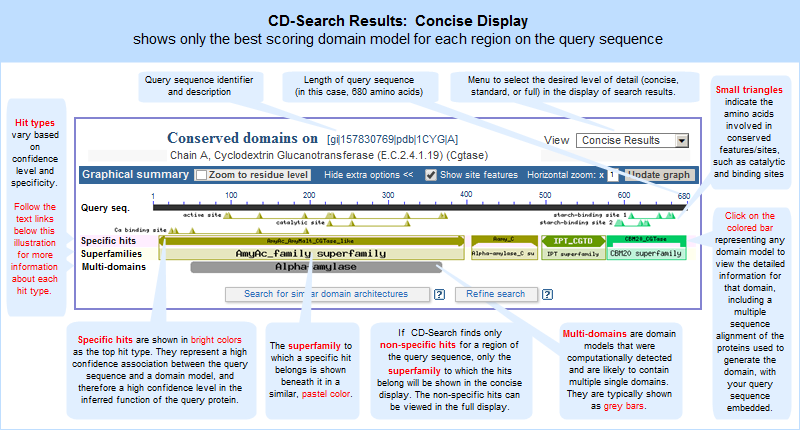
CDD Search Results -- opens a graphical display (illustrated example) of conserved domain model footprints, ranked by their RPS-BLAST score and hit type. A model may appear more than once if it aligns to multiple regions of the query sequence. A concise display showing only the top-scoring hits is presented by default, and it can be changed to a full display of all hits if desired. more...
Conserved Domains (Concise) -- opens a concise list of the conserved domain models that are the top-scoring RPS-BLAST hits to the protein query sequence. Each domain model is listed only once, even if a model had a hit to more than one region on the query sequence. more...
Conserved Domains (Full) -- opens a full list of all the conserved domain models that have a statistically significant RPS-BLAST hit to the protein. Each domain model is listed only once, even if a model had a hit to more than one region on the query sequence. more...
Domain Relatives -- opens a graphical display of similar domain architectures, as determined by the CDART tool, with links to the proteins that have each similar architecture. more...
|
More details about each link are provided in the table below:
| Link Name |
What you will get: |
| CDD Search Results |
The protein sequence → "CDD Search Results" link will open a graphical display, in the CD-Search tool, that shows conserved domain footprints aligned to the protein sequence, based on the results of an RPS-BLAST search of the protein sequence against the PSSMs of all the domain models in the Conserved Domain Database (CDD).
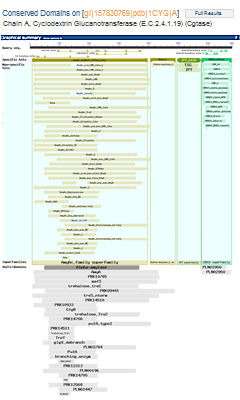
A concise display (depicted below and in a more detailed illustrated example), is shown by default and features only the top scoring models from various hit types. A toggle switch near the upper right corner of the display allows you to see the full display (illustrated example), if desired, which shows all domain models that meet or exceed the RPS-BLAST threshold for statistical significance, i.e., the E-value cutoff. (Open the protein sequence record GI 157830769, featured in the illustration below, and follow the "CDD Search Results" link to try it yourself.)
In both the concise and full views, small triangles indicate the amino acids involved in conserved feaures/sites, such as catalytic and binding sites, when such annotations are available in a domain model.
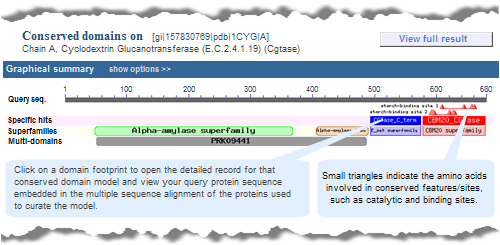
In these graphical views, the domain hits are ordered not only by E-value, but also by classification (specific hits, non-specific hits, superfamilies, multidomains) and model origin (with priority given to NCBI-curated domains if they meet or exceed a domain-specific threshhold score).
If a domain model aligns to more than one region of the query sequence, it will be listed multiple times in the list of domain hits. This is true because the alignment coordinates and score of the domain model vary among different regions of the query sequence, and each hit is reported separately. (The list of domain hits is not shown in the illustration above but is visible beneath the graphical summary on an actual CD-Search results page.)
|
| Conserved Domains (Concise) |
The protein sequence → "Conserved Domains (Concise)" link will open a concise list, in the Entrez Conserved Domain Database, of the conserved domain models that were top-scoring hits to the protein query sequence. Each domain model will be listed only once, even if a model aligned to more than one region on the query sequence.
This list of domains corresponds to those shown in the concise graphical display of CD-Search results.
The same domains will appear in both views, but the order in which they are presented might vary between the displays because the graphical display sorts hits by RPS-BLAST score and hit type, and the list display does not. Also, if a superfamily on the "CDD Search Results" graphical display contains only one domain model, the "Conserved Domains (Concise)" link will retrieve the record for that sole domain model, rather than retrieving the superfamily record.
For example, compare the concise set of top-scoring conserved domains on protein sequence GI 157830769, Cyclodextrin Glucanotransferase, in list vs. graphical format. Note that each link will open in a new window:
Conserved Domains (Concise) -- list format
CDD Search Results -- graphical display
|
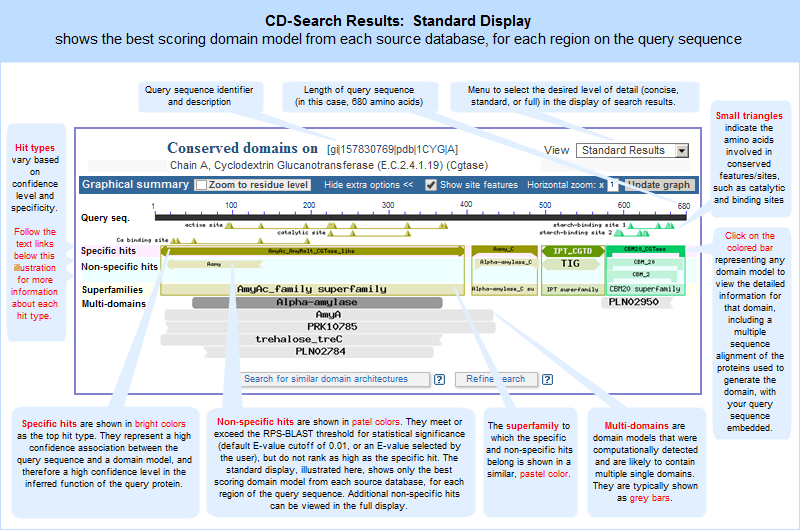
| Conserved Domains (Full) |
The protein sequence → "Conserved Domains (Full)" link will open a full list, in the Entrez Conserved Domain Database, of the all the conserved domain models that have a statistically significant RPS-BLAST hit to the protein query sequence. Each domain model will be listed only once, even if a model aligned to more than one region on the query sequence.
This list of domains corresponds to those shown in the full graphical display of CD-Search results.
The same domains will appear in both views, but the order in which they are presented might vary between the displays because the graphical display sorts hits by RPS-BLAST score and hit type, and the list display does not. Also, if a superfamily on the "CDD Search Results" graphical display contains only one domain model, the "Conserved Domains (Full)" link will retrieve the record for that sole domain model, rather than retrieving the superfamily record.
For example, compare the full set of top-scoring conserved domains on protein sequence GI 157830769, Cyclodextrin Glucanotransferase, in list vs. graphical format. Note that each link will open in a new window:
Conserved Domains (Full) -- list format
CDD Search Results -- graphical display, with "View Full Result" option activated
|
| Domain Relatives |
The protein sequence → "Domain Relatives" link opens a graphical display, in the Conserved Domain Architecture Retrieval (CDART) tool, of similar domain architectures, with links to the proteins that have each similar architecture.
More information about CDART is provided below, in the "domain architecture" section of this document.
|
Domain architecture: CDART:
 The Conserved Domain Architecture Retrieval Tool (CDART) program has been used to analyze the domain architecture of all sequence records in the Entrez Protein database, and to identify proteins with similar architecture. Those proteins are accessible by selecting "Domain Relatives" in the "Links" menu of a protein sequence record of interest (illustrated example).
The Conserved Domain Architecture Retrieval Tool (CDART) program has been used to analyze the domain architecture of all sequence records in the Entrez Protein database, and to identify proteins with similar architecture. Those proteins are accessible by selecting "Domain Relatives" in the "Links" menu of a protein sequence record of interest (illustrated example).
Or, you can search CDART directly by entering a query protein sequence in FASTA format, or entering the GI or Accession number of a protein sequence that already exists in the Entrez Protein database. CDART will then retrieve proteins that contain one or more of the domains present in the query sequence.
More information about CDART is available in the overview, help document, and corresponding publication.
As you are viewing a search results page, click on the thumbnail image or title for any conserved domain model to see it's summary page. The thumbnail will show a snapshot of the 3D structure if a domain model includes a protein sequence(s) from a resolved 3D structure. The thumbnail will depict a sequence alignment if a domain model does not include protein(s) from a resolved 3D structure.
A CD-summary page provides the following information for a domain model (example: cd00400: voltage-gated chloride channel):
- text summary (synopsis of function)
- links to the source database, literature references, and related data in Entrez, as available
- bioassay targets and results, as available
- statistics summarizing salient features of the domain model, such as number of protein sequence rows in the alignment, PSSM identifier, and more
- structure viewing options to display the 3D structure(s), if available, of protein sequences used to curate the domain model.
- conserved features (available for NCBI-Curated domains only)
- sequence cluster phylogenetic tree for protein sequences used to curate the domain (available for NCBI-Curated domains only)
- domain family hierarchy (available for NCBI-Curated domains only)
- multiple sequence alignments of the proteins used to develop the domain model.
More details about each section of the CD-summary page are provided below.
Text Summary (synopsis of function):
- The text summary shown at the top of a CD summary page was written by curators at the source database and provides a synopsis of the domain's biological function. In NCBI curated domains, it also describes the taxonomic extent of the domain, whether it is a monomer or dimer, and any salient features. The text summary in a superfamily record is derived from the representative domain.
Links to related data in Entrez:

- The "Links" box (illustrated at right) on an individual CD Summary page allows you to retrieve additional data associated with the domain model, including related models from the Conserved Domain Database, as well as literature, sequence, and other types of data from across the Entrez system.
- Most of the links are accessible from both the search results page (as "Related information" or "Find Related Data" ads in the right margin) and from the "Links" box on an individual CD Summary page. Links that are present on only one of the pages are noted with an asterisk (*), below. The number and type of links that exist vary among CDD records, depending on the related data that are available for any given record. The link types can include: Related CDs, Literature, Sequence, Structure, BioSystems, and Other Links.
- The "Source" link shown in the illustration opens either: (a) the root (parent) node of an NCBI-curated domain hierarchy, or (b) the corresponding domain record in the external source database, depending upon what type of domain model you are currently viewing. For example, if you are viewing a child or intermediate parent node of a hierarchy, the "Source" link will open the root node. If you are viewing the root node of the hierarchy, the "Source" link will open the corresponding domain model from an external database that was used as a seed for the curated model. If you are viewing the CDD display of a model that was imported from an external database, the "Source" link will open the corresponding domain record on the web site of the source database. In the latter case, note that CDD's display of the model might vary slightly from the source record due to the data processing procedures used by CDD.
|
|
| Link Group |
Link Name * |
Description |
| Related CDs |
Superfamily |
This links to the record for the CDD superfamily to which this domain belongs. |
| Superfamily Members |
This retrieves all the other domain models that belong to the superfamily. |
| Literature |
PubMed |
PubMed citations annotated on the domain. All references have been identified by curators, either by NCBI staff for the NCBI-curated domains, or by the staff of the external databases represented in CDD.
For NCBI-curated domains, the PubMed link leads to the citations that have been annotated on that particular node of a domain family hierarchy, not for all nodes in the tree. Whenever possible, the citations include articles that provide evidence for the biological function of a domain and/or discuss the evolution and classification of a domain family. |
| Free in PMC |
The subset of PubMed links that are available as free full text in PubMed Central. |
| Books * |
Full text information in the Entrez Books database that further clarifies or elucidates the domain's function, the protein's role in metabolic pathways, and other broad overview information, including diagrams and illustrations. |
| Sequence |
Representatives * |
The set of protein sequences that are present in the multiple sequence alignment for the domain model. Following the "Representatives" link will retrieve the complete sequence records from the Entrez Protein database. The number of records retrieved will be identical or similar to the number of Aligned Rows shown in the Statistics box of the CD Summary page. |
| Specific Protein |
The set of protein sequences found by RPS BLAST to contain the domain (with an E-value that is equal to or lower than a domain-specific Threshold E-value). These are called specific hits and represent a very high confidence that the query sequence belongs to the same protein family as the sequences use to create the domain model. The number of proteins you will retrieve by following this link is greater than retrieval from the "Representatives" link, but less than retrieval from "Related Protein" link. |
Related Protein
(Protein †) |
Superset of all protein sequences found by RPS BLAST to contain the domain (with an E-value equal to or better than the default cutoff of 0.01). Therefore, this superset includes two CD-Search hit types: specific hits and non-specific hits. |
Architectures
(Architecture †) |
Proteins found by CDART to contain one or more of the domains present in the proteins that are hit by domains found in the domain superfamily |
| Gene * |
Links from the RPS BLAST concise display hits to the protein sequences listed in Entrez Gene records.
Details: Each protein listed in an Entrez Gene record has been RPS BLASTed against the domain models in CDD. Links are then created between specific regions of those protein sequences and top-scoring domain models which align to them. Top-scoring domain models are shown either as specific-hits, or as the superfamily to which the highest-ranking non-specific hit belongs. |
| HomoloGene * |
Links from the RPS BLAST concise display hits to the protein sequences listed in HomoloGene records. (The details provided for Gene links, above, also apply to HomoloGene links.) |
| Structure |
Related Structure
(Structure †) |
Three-dimensional structures containing at least one protein molecule that has an RPS BLAST hit (specific hit or non-specific hit) to this domain model's PSSM (compare with "Family Structure"). |
| BioSystems |
BioSystems * |
BioSystems containing proteins that have specific hits to the conserved domain. The proteins that have been associated with the BioSystem via the method descibed in data processing/create direct links/proteins.
Note that the BioSystems link can appear in two different places on a search results page:
- The "BioSystems" link in the Find Related Data ad in the right margin of the search results page will retrieve the biosystems associated with all of the conserved domains retrieved by your search, or with those you have selected with checkboxes
- The "BioSystems" link (when present) that is listed beneath an individual conserved domain record on a search results page (or in the "All links for this record" ad in the right margin of the display for an individual record) will open the subset of biosystems containing proteins annotated with that specific domain model. NOTE: If a conserved domain did not get any specific hits to proteins in any BioSystem, it will not have a BioSystem link.
|
| Other Links |
Taxonomy |
The highest node in the NCBI Taxonomy database that encompasses all organisms whose protein sequences are in the multiple sequence alignment for a domain model. The taxonomy link for a superfamily retrieves the highest taxonomic node for all of its constituent domain models. |
| LinkOut * |
External data repositories can link to records in Entrez databases, including the Conserved Domain Database, by using the LinkOut tool. Selecting this option will display brief information about each conserved domain record you have retrieved (such as its accession and short name), followed by the names of (and links to) any external data providers that have chosen to link to the conserved domain (e.g., see cd05582, which has a LinkOut to "Domain Mapping of Disease Mutations"). |
| Family Structure * |
Three-dimensional structures containing at least one protein molecule that has an RPS BLAST specific hit to this domain model's PSSM (compare with "Related Structure"). |
* Most of the links from conserved domain records to related data are accessible from both the search results page (as "Find Related Data" or "Related information" menus) and from the "Links" box on an individual CD Summary page. Links that are present on only one of the pages are noted with an asterisk (*) in the table above, specifically:
The "representatives" and "books" links are found only on the CD summary page for a conserved domain; they are not present in the "Links" menus on a search results page.
Conversely, the "Gene," "HomoloGene," "LinkOut," "Family Structure," and "BioSystems" link currently appear only on the search results page.
† Some links have a short name and a long name. The search results page often uses short names in its "Links" menus due to space limitations, such as "Protein" "Architecture," and "Structure." Those same links are called "Related Protein," Architectures," and "Related Structure," respectively, on an individual CD Summary page.
BioAssay Targets and Results:
-
A section entitled "BioAssay Targets and Results" appears on a conserved domain's summary page only if one or more members of the protein family have been used as targets in PubChem BioAssay records, and if at least one chemical was identified in the experiment(s) to be active against one of the targets defined by the domain family.
As examples:
- view the conserved domain summary page for:
- The "BioAssay Targets and Results" section lists bioassays that have tested the activities of small molecules (e.g., chemicals) against protein sequences that have a specific hit to this domain model, and are therefore considered to be members of this protein family. Some of the information from the bioassays may be generalizable to other members of the protein family, depending on how narrowly a family is defined.
- Up to three representative bioassays are listed as examples in the "BioAssay Targets and Results" box. Click on "Explore more" at the bottom of the box to see a complete list of experiments in the PubChem BioAssay database that have tested small molecules against protein targets belonging to this protein family. From there, you can open the "BioAssays" or "Compounds" folder tabs and click on the counts in the columns such as "active," "inactive," "tested" to see the chemicals that have been screened and their activity potency in the respective bioassays.
Statistics:
| Item |
Description |
| PSSM-ID |
the unique identifier for the position-specific scoring matrix (PSSM) generated by RPS-BLAST for a given multiple sequence alignment. If the sequence alignment changes in any way, for example, if new sequences are added to the alignment, a new PSSM will be generated and will receive a new PSSM-ID.
(Note: Each superfamily record in the Conserved Domain Database also has a PSSM ID, which refers to the specific set of conserved domain PSSM IDs that comprise the superfamily, rather than to an actual position-specific scoring matrix for the overall superfamily.) |
| Aligned |
lists the number of rows in the sequence alignment. In general, each row comes from a different sequence record. However, sometimes two or more rows can be from the same GI number (i.e., same sequence record), if the sequence contains multiple instances of the domain. |
| Threshold Bit Score |
the domain specific threshold score (shown as a bit score) that an RPS-BLAST hit must meet or exceed in order to be considered a specific hit, which represents a high confidence association between a protein query sequence and a conserved domain and therefore a high confidence level for the inferred function of the protein query sequence. The threshold is equal to the weakest E-value (and highest bit score) among self-hits of a domain�s member protein sequences to the resulting domain model (illustrated example). Domain-specific threshold scores are calculated only for NCBI-curated domains. |
| Threshold Setting GI |
the GI number of the member protein sequence (i.e., the protein sequence from the domain model's multiple sequence alignment) that set the threshold bit score. A threshold setting GI number is displayed only for NCBI-curated domains, as thresholds are calculated as part of the curation process. |
| Status |
information about the CD's curation status. Curated models have been realigned by NCBI with consideration of 3D structure. Alignments imported from outside sources have not been changed (except for the import process detailed above) |
| Author |
name of the author who contributed the conserved domain model to the NCBI-curated data set. This line currently appears in records that were contributed by external collaborators (for example, cd08773). Mouse over the name to see a popup with additional contact information, if/as available, for the author. |
| Created |
date at which the seed (or de-novo) alignment was imported into CDD |
| Updated |
date of the most recent changes to the alignment model and/or descriptive information |
Structure:
| Item |
Description |
| "Structure View" Button |
The "Structure View" button in a conserved domain record opens the 3D structure(s), if available, of protein sequences used to curate the domain model. In order for the button to work, the Cn3D program must be installed on your computer. It is a a free helper application available for Windows, Macintosh, and Unix platforms. Installation takes only a couple of minutes and a tutorial describes the program's features and functions.
In addition to displaying an interactive view of the 3D structure(s), Cn3D will also display the multiple sequence alignment of those and other proteins used in the curation of the domain model. The Cn3D structure view and sequence view windows communicate with each other, so highlighting residues in one window will also highlight those residues in the other window.
As noted in the sections on the CD assembly process and unique features of curated domains, NCBI staff include protein sequences from resolved 3D structures (illustration) whenever possible in the multiple sequence alignment of a domain model.
In a multi-level domain hierarchy, the 3D structures might be present in the parent node (e.g., cd00400) if they are not present in an intermediate or terminal node (e.g., cd03683). In that case, click on the parent node to view structures that have been specially annotated to highlight the conserved feature.
You can click on any of the thumbnail structure images on a CD summary page to launch Cn3D. The thumbnail images in the conserved features summary box will launch a specially annotated view of the structure that highlights the particular feature of interest.
However, 3D structures are not always available. If a domain model does not include any structure-based protein sequences, the "Structure View" button will still open Cn3D, but only the sequence viewer window will be populated with data.
Controls in Cn3D will then allow you to manipulate the sequence alignment in various ways, if desired. For example, Cn3D offers column-specific coloring by sequence conservation when invoked with multiple alignment views. This is a convenient feature to study sequence conservation within a CD-alignment and to find out how well the aligned query fits the existing patterns of conservation and variability. The Cn3D tutorial provides more information on the controls available.
|
| Program |
Although the Structure View button provides the option of using an older version of Cn3D (3.0), the default choice is recommended because it uses the most recent public version of the program (currently Cn3D 4.1). |
| Drawing |
Structures, when available, can be displayed in varying levels of detail. All Atoms will load a detailed model. This option transmits a large amount of structure data and loading the structures may therefore take some time. The Virtual Bonds setting displays C-alpha atoms only, with virtual bonds connecting them, and therefore transmits and loads more quickly. |
| Aligned Rows |
By default, Cn3D will display a multiple sequence alignment of up to 10 proteins, starting with sequences whose 3D structures are shown, and then also including sequences from proteins that do not yet have a resolved structure. Use the "aligned rows" menu to increase that number up to 100 rows. |
Conserved Features/Sites summary box (available for NCBI-Curated domains only):
-
If conserved features/sites have been annotated on an NCBI-curated domain, they are noted in a summary box near the top of the page, with one folder tab for each feature (illustrated example).
- Click on the folder tab for a feature of interest to view its details, such as:
- feature number - The ordinal number of the feature within the conserved domain model (e.g., feature 1, feature 2, etc.)
- feature name - Generally reflects the function of the conserved feature/site (e.g., Cl- selectivity filter, Cl- binding residues [ion binding site], dimer interface [polypeptide binding site])
- conserved feature residue pattern - The set of amino acids that characterizes the conserved feature/site.
- The amino acids are not necessarily adjacent to each other in the domain model, but instead appear at the positions designated by hash marks (# symbols) in the multiple sequence alignment of the domain model, as described below.
- The pattern may include ambiguity codes (for example, [ST], which indicates that a position can be occupied by either Serine or Threonine).
- The conserved feature residue pattern is specified by curators, and displayed on a conserved domain summary page, only if it is clear that specific residue types are necessary for the particular molecular function (such as metal coordination, glycosylation attachment, or enzyme catalysis). Therefore, not all conserved features/sites will include a "conserved feature residue pattern" line.
- Note: If a sequence in the Entrez Protein database gets a significant hit to the conserved domain model AND contains the conserved feature residue pattern, the site annotation will be transferred to that protein sequence record.
- evidence - may be free-text comments, literature citations, or "structure evidence" that exemplifies the existence of a site by highlighting an actual molecular complex in an experimentally resolved 3D structure.
- 3D structure thumbnail image, if available. The conserved amino acids that characterize the feature are highlighted in pink, in both the thumbnail image, and in the larger, interactive view of the structure that appears when you click on the thumbnail to launch the structure in the free Cn3D viewing program. (Cn3D must first be loaded on your machine in order for that to work.)
- Clicking the folder tab for a feature of interest will refresh the mutliple sequence alignment display with an extra alignment row that shows the feature number (feature 1, feature 2, etc.) and uses hash-marks (#) to indicate the specific residues involved (also shown in the illustrated example). Only one feature at a time is shown in the multiple sequence alignment display.
Sequence Cluster Phylogenetic Tree (available for NCBI-Curated domains only):
- Based on evidence from sequence comparison, NCBI Conserved Domain Curators attempt to organize related domain models into phylogenetic family hierarchies (details and illustration). Colors used in the sequence cluster phylogenetic tree correspond to colors used in the domain family hierarchy display.
To examine the hierarchical classification more closely, you can download the CDTree program, which is used by the NCBI curators. CDTree enables you to interactively view the complete domain hierarchy, including a detailed display of the sequence cluster tree.
- To view a query protein embedded into the sequence tree of a domain model, first use the CD Search tool to identify the conserved domains in the query sequence. Then click on the cartoon (colored bar) representing a domain of interest in either the Concise Display or Full Display of the CD-Search results page. That will open a CD Summary Page, which shows detailed information about the domain and provides an Interactive Display option for viewing the Hierarchy (an illustrated example is provided in the "How To" pages).
To embed your query in the hierarchy, simply check the box for Add Query Sequence before pressing the "Interactive Display" button. (The free CDTree program must be loaded onto your computer in order for that button to work.) When the CDTree program opens, your query sequence will be highlighted in red. If the sequence tree is large, you might need to de-select the View/Fit to Screen option in CDTree's Sequence Tree window in order see the tree, and the placement of your query sequence, in detail. The CDTree help document is packaged with the software and provides details on how to use the program.
- Algorithms used to generate the cluster diagram in CDTree: The sequence tree viewer in CDTree calculates and displays sequence trees for a set of selected alignment models, which may or may not be linked in a hierarchical fashion. Sequence trees are the graphical depiction of results from simple phylogenetic analysis of the alignment data. Methods available for distance calculation are percent identity, Kimura-corrected percent identity, score of aligned residues, score of optimally extended blocks, blast score for the aligned footprints and blast scores for full-length sequences; a variety of commonly used scoring matrices can be selected. For the sequence trees displayed on CDD web pages, we commonly use "score of aligned residues", where pair-wise alignment scores derived from our multiple sequence alignments, and scored via BLOSUM62, are converted into distances. Trees can be constructed via single-linkage clustering, neighbor joining, or the Fast ME method. We use neighbor-joining for all of the sequence trees displayed on web-pages.
Domain Family Hierarchy (available for NCBI-Curated domains only):
- As noted in the description of NCBI curated domains, the goal of the curation project is to to provide CDD users with insights into how patterns of residue conservation and divergence in a family relate to functional properties. The CD summary page for an NCBI-curated domain shows the hierarchy (details and illustration) to which the currently viewed domain belongs.
Some hierarchies have only one node, while others have many nodes organized into two or more levels. If a hierarchy has multiple nodes, you can click on another node of interest to view the CD summary page for that domain.
Alternatively, you can download the CDTree program used by the NCBI curators in order to view the complete domain hierarchy interactively and in greater detail, with or without a query sequence embedded.
Multiple Sequence Alignment Displays:
- Member proteins used to create domain model:
By default, the sequence alignment display at the bottom of a CD summary page shows 10 of the most diverse members from the cluster of sequences used to create a domain model. (A sample multiple sequence alignment is shown in the illustration of cd00064: Furin-like domain in this help document, or you can open a domain model directly in CDD, such as cd00400: voltage-gated chloride channel.) The multiple sequence alignment display options (below) can be used to change the quantity and appearance of data displayed, and the CD-Search tool can be used if you'd like to embed a query sequence within the alignment.
- Protein query sequence embedded in alignment:
To view a query protein embedded into the multiple sequence alignment of a domain model, first use the CD Search tool to identify the conserved domains in the query sequence. Then click on the cartoon (colored bar) representing a domain of interest in either the Concise Display or Full Display of the CD-Search results page.
- Display Options:
By default, the multiple sequence alignment on a CD summary page is shown in hypertext format and displays up to 10 sequences that were used to curate the domain. The display format, number and type of sequence rows, and color scheme can be changed in the following ways:
| Display Option |
Description |
| Format |
| Hypertext |
Interactive view in which each accession or GI number links to the corresponding complete sequence record in the Entrez Protein database. Displays all residues in each sequence row, with aligned residues shown in upper case, unaligned residues in lower case, and variation in sequence length shown as dashes. A horizontal scale indicates the number of residues in the overall alignment. The numbers at the beginning and end of each sequence row indicate the span of sequence data that was imported from the complete protein sequence record. |
| Plain Text |
This view contains the same content as "Hypertext" but is rendered in ASCII format. |
| Compact Hypertext |
Interactive view in which each accession or GI number links to the corresponding complete sequence record in the Entrez Protein database. Shows only aligned residues (as upper case letters), plus the number of intervening unaligned residues in each sequence row (shown in square brackets []). Does not show the unaligned residues themselves; those are shown only in the "Hypertext" and "Plain Text" format. |
| Compact Text |
This view contains the same content as "Compact Hypertext" but is rendered in ASCII format.
|
| mFASTA |
Multiple FASTA (mFASTA) format is useful for importing the data into sequence analysis programs. For each sequence row in the alignment, it provides a FASTA-formatted definition line ("FASTA defline") followed by up to 80 characters of sequence data on each subsequent line. mFASTA format displays all residues in each sequence row, with aligned residues shown in upper case, unaligned residues in lower case, and variations in length filled in with dashes.
|
|
| Row Display |
Number of rows
in a domain model |
The total number of sequence data rows aligned in a domain model are shown in the statistics portion of that model's CD summary page. |
| Default number shown |
By default, 10 rows of sequence data are shown, including the representative sequence plus nine others. |
| Maximum number shown |
You can change the number of sequence rows displayed using the Row Display pop-up menu. If the Type Selection is set to Most Diverse Members, a maximum of 100 rows can be displayed. If a domain model contains more than 100 rows, the Type Selection Top Listed Sequences allows the display of more than 100 rows. If a model is NCBI-curated, you can also use the CDTree program to view the complete set of rows. Simply install the program, which is free, then press the Interactive Display button in the hierarchy section of the domain model's CD summary page to view all the sequence rows.
Note: In general, each row comes from a different sequence record. However, sometimes two or more rows can be from the same GI number (i.e., same sequence record), if the sequence contains multiple instances of the domain.
|
|
| Type Selection |
| Most Diverse Members |
Lists the representative sequence followed by the most dissimilar protein sequences, as determined from the domain model multiple sequence alignment. They are listed from most to least dissimilar with respect to the representative sequence. |
| Top Listed Sequences |
Merely refers to the order in which the sequences are listed in the multiple alignment; this may or may not be meaningful, depending on the approach used by the source database in curating a particular domain model.
In NCBI-curated domain models, protein sequences from resolved 3-D structures are generally listed first, so the "Top Listed Sequences" display option is useful for bringing these structure-based protein sequences to the top when viewing NCBI-curated domains. The remaining sequences in NCBI-curated domain models are listed in order of increasing GI number or some other non-biological criterion. (This is because the composition of the member sequences, not their order, is important in determining a domain model's position-specific scoring matrix, or PSSM. The other important factor is the degree of residue conservation in any given column of the alignment, which can be visualized with the Color Bits setting, described below.)
The biological relationships among the member sequences of an NCBI-curated domain model are displayed in the sequence cluster phylogenetic tree and the domain family hierarchy on the domain model's CD summary page. Both of these displays can also be viewed interactively using the CDTree program.
|
|
| Color Bits |
| General |
Color Bits allow you to adjust the red <-> blue balance of color used to depict the degree of conservation among aligned (upper case) residues. In general, red indicates highly conserved and blue indicates less conserved. (In other words, the two extremes on the color scale correspond to columns that are completely conserved (e.g., same residue in all alignment rows), and columns with residue types distributed in a way that is no different from the background distribution -- what would seem like a random pick of residue positions from arbitrary protein sequences.) Unaligned (lower case) residues are shown in grey.
The color bit settings can be used to select a threshold for determining which columns are colored in red.
|
Numerical settings |
Higher numbers require higher degrees of conservation within an alignment column (i.e., less residue variation) in order to display that column in red font.
The score threshold that must be met in order for an alignment column to be displayed in red can be adjusted from a low of 0.5 to a high of 4.0. As the threshold increases, the number of columns shown in red will decrease.
Background: Each column in the multiple sequence alignment display receives a score that indicates that column's "information content" -- its contribution to the overall alignment score -- indicating how important the column is as an "anchor" for the alignment. The higher the score, the more important that column is in the alignment.
We use a fairly standard definition of "information content" for an aligned column:
SUM (f(i) * log (f(i)/q(i))
over all base 2
residue
types i
where f(i) is the observed relative residue frequency, and q(i) is the background/reference relative frequency for that residue type (based on the table that accompanies the BLOSUM62 matrix). This is also called "relative entropy", which is a popular way to measure the distributions of nucleotide bases or amino acids.
A column's score is calculated on the fly, based on the sequence rows currently shown in the display. As the number and type of sequence rows in the display change, the column's score, and therefore its color, can change.
|
| Identity setting |
The Identity setting uses red font only in columns that contain the same residue in all of the sequence rows displayed. All other aligned columns are colored in blue and unaligned columns are shown in grey.
|
|
| "Feature" hash marks (#) |
Hash-marks (#) in the top row of a multiple sequence alignment display indicate the specific residues involved in a conserved feature, such as a binding or catalytic site, that has been annotated on an NCBI-curated domain.
Although multiple features may have been annotated, only one feature at a time is shown in the multiple sequence alignment display.
A conserved features/sites summary box (illustration) lists the features that have been annotated. Clicking on the tab for a feature of interest will show its details. It will also refresh the mutliple sequence alignment display to mark the residues involved in the currently viewed feature (as depicted in the bottom of the illustration).
|
CDD is updated several times a year. We no longer try to follow updates of the source databases on a regular basis,
but will re-import source database content occasionally. CDD continues to grow, however, through NCBI's curation effort.
At the moment, CDD curators focus on capturing and describing hierarchies of related domain families, which are, for the
most part, covered by the imported un-curated models as well. The current curation effort is restricted to ancient domain families with wide phylogenetic distribution, and focuses on families with at least one 3D structure representative.
The scientific community's understanding of molecular data continues to evolve as research progresses. Some domain models in CDD are generated through automated processes and others are curated. All are fluid and revised as new data become available and as new protein family clustering methods are developed. Because of this, we welcome your feedback on the data at [email protected], including information/annotations you find particularly helpful as well as any discrepancies you may notice.
|