Summary
Clinical characteristics.
While the majority of individuals with HRAS-related Costello syndrome (Costello syndrome) share characteristic findings affecting multiple organ systems, the phenotypic spectrum is wide, ranging from a mild or attenuated phenotype to a severe phenotype with early-lethal complications. Costello syndrome is typically characterized by failure to thrive in infancy as a result of severe postnatal feeding difficulties; short stature; developmental delay or intellectual disability; coarse facial features (full lips, large mouth, full nasal tip); curly or sparse, fine hair; loose, soft skin with deep palmar and plantar creases; papillomata of the face and perianal region; diffuse hypotonia and joint laxity with ulnar deviation of the wrists and fingers; tight Achilles tendons; and cardiac involvement including cardiac hypertrophy (usually hypertrophic cardiomyopathy), congenital heart defects (usually valvular pulmonic stenosis), and arrhythmia (usually supraventricular tachycardia, especially abnormal atrial rhythm / multifocal atrial tachycardia or ectopic atrial tachycardia). Relative or absolute macrocephaly is typical, and postnatal cerebellar overgrowth can result in the development of a Chiari I malformation with associated anomalies including hydrocephalus or syringomyelia. Individuals with Costello syndrome have an approximately 15% lifetime risk for malignant tumors including rhabdomyosarcoma and neuroblastoma in young children and transitional cell carcinoma of the bladder in adolescents and young adults.
Diagnosis/testing.
The diagnosis of Costello syndrome is established in a proband with suggestive clinical findings and a heterozygous HRAS pathogenic variant identified by molecular genetic testing.
Management.
Targeted therapy: Trametinib (MEK inhibitor) for treatment of hypertrophic cardiomyopathy with heart failure that is refractory to standard treatment.
Supportive care: Failure to thrive is the most common and challenging clinical problem; most infants require nasogastric or gastrostomy feeding, and many require Nissen fundoplication. Cardiac manifestations and malignancies are managed through standard protocols. Ulnar deviation of the wrists and fingers often requires early bracing and occupational and/or physical therapy; tight Achilles tendons may require surgical tendon lengthening. Developmental delay requires early intervention programs and individualized education strategies. Recurrent facial papillomata may require removal. General anesthesia may pose a risk to those with hypertrophic cardiomyopathy or those predisposed to types of atrial tachycardia.
Surveillance: Monitoring for neonatal hypoglycemia; echocardiography with electrocardiogram at the time of diagnosis with subsequent follow up by a cardiologist; abdominal and pelvic ultrasound examinations to screen for rhabdomyosarcoma and neuroblastoma every three to six months until age eight to ten years may be considered; annual urinalysis for evidence of hematuria to screen for bladder cancer beginning at age ten years.
Genetic counseling.
Costello syndrome is an autosomal dominant disorder typically caused by a de novo pathogenic variant. Rarely, an individual with Costello syndrome has the disorder as the result of an HRAS pathogenic variant inherited from a heterozygous parent; vertical transmission has been reported in two families with the rare, attenuated phenotype of Costello syndrome. If the HRAS pathogenic variant identified in the proband cannot be detected in the leukocyte DNA of either parent, the recurrence risk to sibs is slightly greater than that of the general population because of the possibility of parental germline mosaicism. Recurrence of Costello syndrome in sibs has been reported and is suspected to be the result of germline mosaicism in a parent. Individuals with classic Costello syndrome typically do not reproduce. If an HRAS pathogenic variant has been identified in an affected family member, prenatal and preimplantation genetic testing are possible.
Diagnosis
No consensus clinical diagnostic criteria for HRAS-related Costello syndrome (Costello syndrome) have been published to date.
Suggestive Findings
Costello syndrome should be suspected in probands with the following clinical and neuroimaging findings and family history.
Clinical Findings
Prenatal findings
- On ultrasound examination:
- Increased nuchal thickness
- Polyhydramnios (>90%)
- Characteristic ulnar deviation of the wrists
- Short humeri and femurs
- Fetal tachycardia (various forms of atrial tachycardia)
- Preterm delivery
Postnatal findings
- Severe postnatal feeding difficulties extending throughout early childhood
- Failure to thrive
- Short stature
- Macrocephaly (relative or absolute)
- Curly or sparse, fine hair

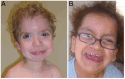
Figure 2.
Typical facial features seen in a boy age eight years of northern European background (A) and a Hispanic girl age ~11 years (B) with Costello syndrome Reprinted with permission from Gripp & Lin [2012]
Skin
- Loose, soft skin
- Increased pigmentation
- Deep palmar and plantar creases
- Papillomata of face and/or perianal region (typically absent in infancy but may appear in childhood)
- Hyperkeratosis and calluses
- Premature aging with hair loss
Musculoskeletal system
- Diffuse hypotonia, joint laxity, and low muscle mass
- Ulnar deviation of wrists and fingers; splayed fingers resulting in characteristic hand posture
- Spatulate finger pads, abnormal fingernails
- Tight Achilles tendons (often evolving throughout childhood)
- Positional foot deformity
- Vertical talus
- Kyphoscoliosis
- Pectus carinatum, pectus excavatum, asymmetric rib cage
- Developmental hip dysplasia
Cardiovascular system
- Cardiac hypertrophy, usually hypertrophic cardiomyopathy (i.e., idiopathic subaortic stenosis, asymmetric septal hypertrophy), although other forms (e.g., biventricular hypertrophy) have been reported
- Congenital heart defects, usually valvular pulmonic stenosis
- Arrhythmia, usually supraventricular tachycardia. Most distinctive is chaotic atrial rhythm / multifocal atrial tachycardia or ectopic atrial tachycardia (known as non-reentrant tachycardias)
- Aortic dilatation (typically mild, noted in fewer than 10% of individuals)
- Hypertension
Neurologic
- Chiari I malformation (may develop over time)
- Hydrocephalus
- Syringomyelia
- Tethered cord
- Seizures
Tumors. Increased occurrence of malignant solid tumors including rhabdomyosarcoma and neuroblastoma in young children and transitional cell carcinoma of the bladder in adolescents and young adults
Psychomotor development
- Developmental delay or intellectual disability
- Findings suggestive of autism spectrum disorder in early infancy (that typically improve by age four years)
- Sociable, outgoing personality
- Anxiety
Neuroimaging findings
- Posterior fossa crowding with cerebellar tonsillar ectopia or herniation
- Tethered cord
- Ventriculomegaly or hydrocephalus
Family History
Because Costello syndrome is typically caused by a de novo pathogenic variant, most probands represent a simplex case (i.e., a single occurrence in a family).
Establishing the Diagnosis
The clinical diagnosis of Costello syndrome can be established in a proband based on suggestive physical exam and imaging findings, or the molecular diagnosis can be confirmed in a proband with a heterozygous pathogenic (or likely pathogenic) variant in HRAS [Aoki et al 2005, Kerr et al 2008, Grant et al 2018] (see Table 1).
Note: (1) Per ACMG/AMP variant interpretation guidelines, the terms "pathogenic variant" and "likely pathogenic variant" are synonymous in a clinical setting, meaning that both are considered diagnostic and can be used for clinical decision making [Richards et al 2015]. Reference to "pathogenic variants" in this GeneReview is understood to include likely pathogenic variants. (2) Identification of a heterozygous HRAS variant of uncertain significance does not establish or rule out the diagnosis.
Molecular genetic testing approaches can include a combination of gene-targeted testing (single-gene testing, multigene panel) and comprehensive genomic testing (exome sequencing, genome sequencing) depending on the phenotype. Gene-targeted testing requires that the clinician determine which gene(s) are likely involved (see Option 1), whereas genomic testing does not (see Option 2).
Option 1
When the clinical findings suggest the diagnosis of Costello syndrome, molecular genetic testing approaches can include single-gene testing or use of a multigene panel.
- Single-gene testing. Sequence analysis of HRAS is performed first to detect missense variants and small intragenic deletions/insertions. Note: Depending on the sequencing method used, single-exon, multiexon, or whole-gene deletions/duplications may not be detected. Note: If no variant is detected by the sequencing method used, the next step is to perform gene-targeted deletion/duplication analysis to detect exon and whole-gene deletions or duplications; however, to date such variants have not been identified as a cause of Costello syndrome, as it is caused by activating single-nucleotide variants in HRAS.
- A multigene panel that includes HRAS and other genes of interest (see Differential Diagnosis) is most likely to identify the genetic cause of the condition while limiting identification of variants of uncertain significance and pathogenic variants in genes that do not explain the underlying phenotype. Note: (1) The genes included in the panel and the diagnostic sensitivity of the testing used for each gene vary by laboratory and are likely to change over time. (2) Some multigene panels may include genes not associated with the condition discussed in this GeneReview. (3) In some laboratories, panel options may include a custom laboratory-designed panel and/or custom phenotype-focused exome analysis that includes genes specified by the clinician. (4) Methods used in a panel may include sequence analysis, deletion/duplication analysis, and/or other non-sequencing-based tests.
Option 2
When the diagnosis of Costello syndrome is not considered because an individual has atypical phenotypic features, comprehensive genomic testing does not require the clinician to determine which gene is likely involved. Exome sequencing is most commonly used; genome sequencing is also possible. To date, the majority of reported HRAS pathogenic variants are within the coding region and are likely to be identified by exome sequencing.
For an introduction to comprehensive genomic testing click here. More detailed information for clinicians ordering genomic testing can be found here.
Table 1.
Molecular Genetic Testing Used in Costello Syndrome
Clinical Characteristics
Clinical Description
HRAS-related Costello syndrome (Costello syndrome) affects multiple organ systems. The typical presentation is characterized by diffuse hypotonia and severe feeding difficulties in infancy; short stature; developmental delay or intellectual disability; characteristic facial features; curly or sparse, fine hair; loose, soft skin with deep palmar and plantar creases; papillomata of the face and perianal region; joint laxity with ulnar deviation of the wrists and fingers; tight Achilles tendons; and cardiac involvement (including hypertrophic cardiomyopathy [HCM], congenital heart defects, and arrhythmia). Postnatal cerebellar overgrowth can result in Chiari I malformation with associated hydrocephalus or syringomyelia. There is an approximate 15% lifetime risk for malignant tumors in individuals with Costello syndrome including rhabdomyosarcoma and neuroblastoma in young children and transitional cell carcinoma of the bladder in adolescents and young adults.
To date, more than 100 individuals have been identified with a pathogenic variant in HRAS [Aoki et al 2005, Estep et al 2006, Gripp et al 2006a, Kerr et al 2006, van Steensel et al 2006, Lin et al 2011, Weaver et al 2014]. The following description of the phenotypic features associated with this condition is based on these reports.
Table 2.
Costello Syndrome: Frequency of Select Features
Growth. Increased birth weight and head circumference (often >50th centile) for gestational age can lead to the categorization of Costello syndrome as a macrosomia disorder, which is misleading. Increased birth weight is instead related to fetal hydrops. Short stature is universal, delayed bone age is common [Johnson et al 1998], and testing may show partial or complete growth hormone deficiency.
Failure to thrive and severe feeding difficulties are almost universal and typically require a gastrostomy tube. Anecdotally, affected children have very high caloric needs. Even after nutrition is improved through supplemental feeding, growth restriction persists; therefore, aggressive (hypercaloric) feeding therapy is not effective. Children are typically able to take oral feeds beginning between ages two and four years.
Normative growth charts, derived from measurements of individuals who had not used growth hormone, document very slow weight gain in early infancy as well as short stature, with the 95th centile for individuals with Costello syndrome falling into the low-normal range of typical age-matched individuals [Sammon et al 2012]. The reported adult height range is 135-150 cm [Hennekam 2003].
Neurologic. Most infants have hypotonia, irritability, developmental delay, and nystagmus.
Hypotonia may be severe with low muscle mass and a skeletal myopathy phenotype [van der Burgt et al 2007, Tidyman et al 2011].
Progressive postnatal cerebellar overgrowth may result in the development of Chiari I malformation, syringomyelia, and hydrocephalus [Gripp et al 2010]. Cerebellar abnormalities include tonsillar ectopia or Chiari malformation, occasionally associated with syringomyelia [Gripp et al 2000, Gripp et al 2002, Calandrelli et al 2015].
EEG abnormalities are seen in approximately one third of individuals; between 20% and 50% have seizures [Delrue et al 2003, Kawame et al 2003].
Cardiac abnormalities, which typically present in infancy or early childhood, may be recognized at any age. In 146 individuals with molecularly confirmed Costello syndrome, 87% had some type of cardiovascular abnormality. A congenital heart defect was present in 44%, with non-progressive valvular pulmonic stenosis being the most common finding. Rarely, atrial septal defects are seen. Hypertrophic cardiomyopathy (HCM) comprising typical subaortic septal hypertrophy was noted in 61% and pathologic myocardial disarray was seen in 70% of those studied [Lin et al 2011].
A few neonates can present with very severe HCM that is lethal. In other infants, progressively severe HCM and/or severe multifocal atrial tachycardia can lead to death in the first two years of life. Use of the MEK inhibitor trametinib may be considered in these individuals [Geddes et al 2023] (see Targeted Therapy). Multifocal atrial tachycardia and other types pf atrial tachycardia may be very concerning but are usually self-limited with aggressive treatment.
Pulmonic valve stenosis is usually mild to moderate, and infrequently requires surgery or interventional catheterization.
Most children with HCM have either mild or moderate involvement. Of great interest are the few with moderate-to-severe involvement who appear to have "remodeling" over many years that gives the impression of disappearance of (or marked decrease in) left ventricular obstruction. Only a small number of these individuals are being followed, and their long-term natural history is incomplete [Lin et al 2011]. In addition to the rare severe lethal form, HCM can be chronic (persistent in its gradient severity) or progressive (increasing in gradient severity; 14/37 [37%]), stabilizing (without further increase in severity; 10/37 [27%]), or decreasing (resolving; 5/37 [14%]). Outcome was unavailable in 8/37 (22%) [Lin et al 2011], necessitating prudent surveillance.
Non-reentrant atrial tachycardias are generally self-limited but may persist or worsen in approximately one fourth of affected individuals. Non-reentrant atrial tachycardia occurs independently of HCM [Levin et al 2018].
Older individuals (ages 16 to 40 years) with moderate HCM or new-onset arrhythmia (both atrial and ventricular) represent the greatest challenge, and the outcomes in these individuals are not known. Hypertension is not uncommon.
Mild-to-moderate aortic dilatation not associated with bicuspid aortic valve is reported in approximately 5% of affected individuals [Lin et al 2011].
Primary vascular disease has rarely been reported.
Developmental delay or intellectual disability is present in all individuals [Axelrad et al 2004, Axelrad et al 2007, Axelrad et al 2009, Axelrad et al 2011].
Recognition memory in verbal memory functioning is relatively preserved compared to other cognitive tasks [Schwartz et al 2013].
While the underlying mechanism in Costello syndrome is not known, the skills necessary for swallowing and speech development are both affected and appear closely correlated. The onset of speech frequently coincides with the willingness to feed orally.
Behavioral/social issues. Many children younger than age four years meet criteria for autism spectrum disorder (ASD). In contrast, none of the children older than age four years met criteria for ASD, suggesting that early signs consistent with ASD tend to resolve by age four years [Schwartz et al 2017].
Separation anxiety, seen in 39% of individuals with Costello syndrome, is more common in males than in females [Axelrad et al 2011]. In older individuals, anxiety was reported in 65% [Shikany et al 2020].
Limited detailed information is available on the quality of life in older individuals with Costello syndrome. Quality of life in individuals aged 16-34 years is compromised by four factors: limited relationships outside of the immediate circle of friends and family, lack of independence, male sex, and the presence of major medical issues [Hopkins et al 2010]. Functional limitations from orthopedic issues related to mobility, as well as limitations in social and cognitive domains, were documented using normative scales [Johnson et al 2015]. A query of 20 individuals aged 16 years or older found one individual living independently, three living in a residential setting, and 16 living with family [Shikany et al 2020]. A history of seizures was reported in five (25%) and Chiari I malformation in 10 (50%). Mobility issues included the use of a walker in one (5%), wheelchair or stroller for distance only in nine (45%), wheelchair reliance in three (15%), and no mobility issues in seven individuals (35%) [Shikany et al 2020].
Dermatologic. Papillomata, absent in infancy, appear in young children, usually in the perinasal region and less commonly in the perianal region, torso, and extremities. While papillomata are mostly of cosmetic concern, they can become noticeable and at times bothersome.
Palmoplantar keratoderma is common and can affect function in severe cases [Marukian et al 2017]. Additional findings include acanthosis nigricans and thick toenails.
Musculoskeletal. Individuals with Costello syndrome have very loose joints, particularly involving the fingers. Ulnar deviation of the wrists and fingers is also common. Developmental hip dysplasia may result in severe pain and prevent ambulation. Tight Achilles tendons may develop and were either present or previously surgically corrected in 11 of 17 individuals (65%) [Leoni et al 2021].
More than half of a cohort of 43 individuals examined by an orthopedic surgeon with review of available radiographs showed ligamentous laxity, scoliosis, kyphosis, characteristic hand and wrist deformities, shoulder and elbow contractures, tight Achilles tendons, and flat feet [Detweiler et al 2013]. Hip dysplasia, seen in 45% of individuals, was not universally congenital but acquired in some.
Osteoporosis is common in young adults with Costello syndrome [White et al 2005, Detweiler et al 2013]. In adults ranging in age from 16 to 40 years, all eight individuals who had bone density measurements had abnormal results that suggested osteoporosis or osteopenia; three had bone pain, vertebral fractures, and height loss [White et al 2005]. In a study of nine individuals with Costello syndrome who had dual-energy x-ray absorptiometry scans performed, all showed significantly decreased bone mineral density compared to age-matched controls [Leoni et al 2014]. A review of older individuals identified osteoporosis or osteopenia in 9/20 (45%) and kyphosis or scoliosis in 15/20 (75%) [Shikany et al 2020].
Respiratory. Seven of ten individuals ages three to 29 years who underwent polysomnography had obstructive sleep apnea [Della Marca et al 2006]. A literature review showed respiratory complications in 78% of neonates, with the majority resolving over time [Gomez-Ospina et al 2016].
More severe complications (intrauterine hydrops, postnatal pulmonary effusions with respiratory compromise, and severe progressive HCM) have been reported in a few individuals with HRAS pathogenic variants associated with the severe Costello syndrome phenotype [Gomez-Ospina et al 2016] (see Genotype-Phenotype Correlations).
Upper airway obstruction was seen more often in older children and young adults [Gomez-Ospina et al 2016].
Endocrine. Neonatal hyperinsulinism has been reported [Alexander et al 2005, Sheffield et al 2015] and in one case was correlated with focal uniparental disomy for 11p within the pancreatic nodule [Gripp et al 2016].
In older individuals, hypoglycemia may be related to growth hormone deficiency. Growth hormone deficiency is common (30%-50%) [Estep et al 2006, Gripp et al 2010].
Several affected individuals have been diagnosed with hypothyroidism requiring thyroid hormone replacement.
Other endocrine issues may include delayed or dysregulated puberty including precocious puberty.
Solid tumors. Benign and malignant solid tumors occur with far greater frequency in individuals with Costello syndrome than the general population. The overall tumor incidence is approximately 15% over the lifetime of individuals with HRAS pathogenic variants [Gripp et al 2006a]. Kratz et al [2011] reviewed published cases and confirmed the 15% cumulative incidence of cancer in individuals with Costello syndrome by age 20 years. Rhabdomyosarcoma occurs most frequently, followed by neuroblastoma, transitional cell carcinoma of the bladder, and other solid tumors [Gripp 2005].
In a meta-analysis of 234 publications reporting 621 individuals from 35 countries, more than 9% had cancer, including rhabdomyosarcoma, transitional cell carcinoma of the bladder, and neuroblastoma. Cumulative incidence by age 20 years was 13% for cancer and 11% for cancer-free death. Death rate was 3%-4% until age three years. Survival after cancer appeared reduced [Astiazaran-Symonds et al 2023].
Rhabdomyosarcoma and neuroblastoma, tumors of early childhood, present in Costello syndrome at ages comparable to the general population. In contrast, transitional cell carcinoma of the bladder, which occurs in older adults (70% occurs in adults age >65 years) in the general population, may be found in adolescents with Costello syndrome. The ages at presentation in the three individuals with Costello syndrome with transitional cell carcinoma of the bladder were ten, 11, and 16 years. A review of cystoscopy findings in 13 individuals aged ten years or older found a macroscopic bladder lesion in 10/13 on first exam. Histology showed low-grade epithelial dysplasia in 7/10 (70%) and papillary urothelial neoplasm of low-malignant potential (PUNLMP) or low-grade bladder cancer in 3/10 (30%) [Leoni et al 2022].
Other
- Pyloric stenosis occurs more commonly in Costello syndrome than in the general population [Gripp et al 2008].
- Craniosynostosis of the sagittal and/or coronal sutures requiring surgical repair has been reported in several individuals [Weaver et al 2022].
- Cryptorchidism is frequent, structural ureteral or renal abnormalities may be found, and urachal remnants have been noted in multiple individuals [Myers et al 2014, Lorenz et al 2012].
- Adult-onset gastroesophageal reflux was present in four individuals in White et al [2005]; additional cases are known [K Gripp, personal observation].
- Dental abnormalities, including enamel defects, occur frequently. Malocclusion with maxillary first molars positioned posteriorly to the mandibular first molars is common and may contribute to obstructive sleep apnea [Goodwin et al 2014]. Excessive secretions are often noted [Johnson et al 1998].
- In addition to the common vision disturbance and nystagmus, less common eye abnormalities include retinal dystrophy [Pierpont et al 2017] and keratoconus [Gripp & Demmer 2013].
- Adolescents may appear older than their chronologic age because of worsening kyphoscoliosis, sparse hair, and prematurely aged skin.
Neuroimaging. A systematic review of brain and spinal cord MRI studies revealed posterior fossa crowding with cerebellar tonsillar herniation in 27/28 (96%) individuals [Gripp et al 2010]. In a majority of those with serial studies, posterior fossa crowding progressed. Due to the progressive nature of the cerebellar overgrowth – which likely results from abnormal cell differentiation as reported by Paquin et al [2009] – the sequelae of posterior fossa crowding included hydrocephalus requiring shunt placement or ventriculostomy (7/28), Chiari I malformation (9/28), and syringomyelia (7/28). In individuals age 16 years or older, a Chiari I malformation was reported in 10/20 (50%) individuals [Shikany et al 2020].
Tethered cord is relatively common.
Life expectancy. Causes of death in the literature, including an analysis of cardiovascular findings [Lin et al 2011], were reported in 10% of affected individuals and include HCM, coronary artery fibromuscular dysplasia, multifocal tachycardia, neoplasia, pulmonary causes, and multiorgan failure.
Mosaic Costello syndrome. Individuals with somatic mosaicism for a pathogenic variant in HRAS may show patchy skin findings only (as reported in the father of an individual with typical Costello syndrome [Sol-Church et al 2009, Bertola et al 2017]) or have findings indistinguishable from Costello syndrome caused by a germline pathogenic variant [Girisha et al 2010].
One individual with somatic mosaicism (20%-30% of DNA derived from buccal cells had the pathogenic HRAS variant p.Gly12Ser, which was not detected in DNA derived from peripheral blood cells) had typical findings attributed to Costello syndrome (intellectual disability, short stature, sparse hair, coarse facial features, nasal papillomata, and tight Achilles tendons) as well as several atypical findings attributed to mosaicism (microcephaly, streaky areas of skin hypo- and hyperpigmentation, and normal menarche with subsequent regular menses) [Gripp et al 2006b].
Genotype-Phenotype Correlations
In a systematic review of 146 individuals with an HRAS pathogenic variant, there was no apparent correlation between the specific variant and the variables studied (HCM, multifocal tachycardia, aortic dilatation) [Lin et al 2011].
Because few affected individuals with HRAS pathogenic variants other than p.Gly12Ser have been identified, limited genotype-phenotype correlations have been observed:
- The p.Gly13 amino acid appears to be the second most commonly affected residue, with p.Gly13Cys being the most frequent pathogenic variant seen at this codon. Gripp et al [2011a] reviewed physical findings in 12 individuals with this pathogenic variant and noted a distinctive phenotype including dolichocilia (extremely long eye lashes, often requiring trimming) and loose anagen hair syndrome; neither of these findings had previously been noted in individuals with Costello syndrome. Papillomata or multifocal atrial tachycardia have not been seen to date in individuals with p.Gly13Cys. Compared to individuals with the very common p.Gly12Ser pathogenic variant, these differences are statistically significant.
- Kerr et al [2006] suggested that the risk for malignant tumors may be higher in individuals with the p.Gly12Ala pathogenic variant (4/7) than those with the p.Gly12Ser pathogenic variant (4/65). No individuals with p.Gly13Cys have developed a malignant tumor to date [Gripp et al 2011a].
- The suggestion of Lo et al [2008] that a more severe neonatal phenotype may be associated with certain rare pathogenic variants, including p.Gly12Ala and p.Gly12Cys, was confirmed by McCormick et al [2013].
- The possibility of a milder or attenuated phenotype was noted in individuals with the pathogenic variants p.Thr58Ile and p.Ala146Val [Gripp et al 2008], as well as p.Gly60Val [Gripp et al 2017].
- Two unrelated individuals with p.Glu37dup shared phenotypic findings including very sparse hair and facial features that appear less coarse than most other individuals with Costello syndrome [Gremer et al 2010].
- Five individuals with the rare p.Gly13Asp pathogenic variant showed an apparently milder presentation and none had a malignancy [Bertola et al 2017].
In rare instances related to the underlying pathogenic HRAS variant, the presentation is more severe, with intrauterine hydrops, postnatal pulmonary effusions with respiratory compromise, and severe progressive HCM, resulting in early lethality [Lo et al 2008]. Other rare pathogenic variants, such as p.Thr58Ile and p.Gly60Val, are associated with a milder or attenuated phenotype, encompassing milder developmental delay, less striking facial features resembling Noonan syndrome, and a lower risk of malignancy [Gripp et al 2015, Bertola et al 2017].
In one individual with early-lethal Costello syndrome due to the rare p.Gly12Glu pathogenic variant, pulmonary vascular dysplasia affecting the small arteries and veins with abnormal elastin distribution was seen in the absence of significant HCM [Weaver et al 2014].
In a recent meta-analysis of cancer risk in Costello syndrome, the rate of cancer and death associated with p.Gly12Ser were lower when compared to all other pathogenic variants affecting the same amino acid (tumor risk assessed to be 9.2% for p.Gly12Ser, 38.7% for p.Gly12Ala, and 37.5% for p.Gly12Cys) (P <0.05). Higher mortality for p.Gly12Cys, p.Gly12Asp, p.Gly12Val, and p.Gly60Val and higher malignancy rate for p.Gly12Ala were confirmed (P <0.05) [Astiazaran-Symonds et al 2023].
Penetrance
Penetrance for Costello syndrome is 100% to date [Aoki et al 2005, Estep et al 2006, Gripp et al 2006a, Kerr et al 2006].
Nomenclature
Costello reported the first individuals with this condition in 1971, providing follow up in 1977 and 1996 [Costello 1971, Costello 1977, Costello 1996]. The eponym was applied for the first time by Der Kaloustian et al [1991].
Early examples of Costello syndrome were reported as:
- AMICABLE syndrome (amicable personality, mental retardation, impaired swallowing, cardiomyopathy, aortic defects, bulk, large lips and lobules, ectodermal defects) [Hall et al 1990];
- Faciocutaneous-skeletal syndrome [Borochowitz et al 1992].
Prevalence
The birth prevalence of Costello syndrome is estimated to be 1:380,000 in the United Kingdom [Giannoulatou et al 2013]. In contrast, an epidemiologic study using national surveys suggested a prevalence of 1:1,230,000 in Japan [Abe et al 2011]; this may be an underestimate due to ascertainment bias.
Genetically Related (Allelic) Disorders
Germline pathogenic variants. No phenotypes other than those discussed in this GeneReview are known to be associated with germline pathogenic variants in HRAS.
Somatic mosaicism. De novo postzygotic genetic alterations in HRAS resulting in somatic mosaicism have been reported in individuals with the following:
- Schimmelpenning-Feuerstein-Mims syndrome (also referred to as linear sebaceous nevus syndrome [OMIM 163200]) – consisting of a linear nevus sebaceous with extracutaneous anomalies affecting the brain, eyes, or skeleton – is caused by somatic HRAS pathogenic variants, most often c.37G>C (p.Gly13Arg), and rarely other mosaic pathogenic variants affecting the residues p.Ala11 or p.Gly12 [Groesser et al 2012, Ono et al 2023].
- Cutaneous-skeletal hypophosphatemia syndrome consists of ectodermal findings including epidermal nevi and elevated fibroblast growth factor 23 with hypophosphatemia and can be due to mosaic HRAS pathogenic variants including p.Gly13Arg [Lim et al 2016].
Sporadic tumors (including solid tumors of adulthood, such as bladder carcinoma or lung carcinoma) occurring as single tumors in the absence of any other findings of Costello syndrome frequently contain a somatic pathogenic variant in HRAS that is not present in the germline. In these circumstances predisposition to these tumors is not heritable. Somatic mutational hot spots include the glycines at amino acid residues 12 and 13 and the glutamine at residue 61. Pathogenic missense variants in these codons lead to increased activity of the gene product.
Differential Diagnosis
No other loci have been identified as causative of HRAS-related Costello syndrome (Costello syndrome) [Grant et al 2018]. In earlier series, the 10%-15% of individuals suspected of having Costello syndrome who lacked an HRAS pathogenic variant were subsequently found to have cardiofaciocutaneous (CFC) syndrome [Rauen 2006, Gripp et al 2007] or pathogenic variants in KRAS typical of Noonan syndrome [Lo et al 2009].
Note: While Costello syndrome is difficult to distinguish from CFC syndrome or Noonan syndrome in infants and young children, the distinction between Costello syndrome and Noonan syndrome is clearer in older children.
Table 3.
Disorders of Interest in the Differential Diagnosis of Costello Syndrome
Management
Clinical practice guidelines for the management of HRAS-related Costello syndrome (Costello syndrome) have been published [Gripp et al 2019].
Evaluations Following Initial Diagnosis
To establish the extent of disease and needs in an individual diagnosed with Costello syndrome, the evaluations summarized in Table 4 (if not performed as part of the evaluation that led to the diagnosis) are recommended.
Table 4.
Costello Syndrome: Recommended Evaluations Following Initial Diagnosis
Treatment of Manifestations
Targeted Therapy
In GeneReviews, a targeted therapy is one that addresses the specific underlying mechanism of disease causation (regardless of whether the therapy is significantly efficacious for one or more manifestation of the genetic condition); would otherwise not be considered without knowledge of the underlying genetic cause of the condition; or could lead to a cure. —ED
Table 5.
Costello Syndrome: Targeted Therapy
Supportive Care
There is no cure for Costello syndrome. Supportive care to improve quality of life, maximize function, and reduce complications is recommended. This ideally involves multidisciplinary care by specialists in relevant fields (see Table 6).
Table 6.
Costello Syndrome: Treatment of Manifestations
Growth hormone (GH) treatment. If treatment with GH is contemplated, its unproven benefit and potential risks should be thoroughly discussed in view of the established risks of cardiomyopathy and malignancy in individuals with Costello syndrome. GH replacement has not been shown to increase these risks.
- Unproven benefit. Individuals with Costello syndrome frequently have low GH levels.
- True GH deficiency requires GH replacement. Three individuals with GH deficiency showed increased growth velocity without adverse effects after three to seven years of replacement therapy, but two continued to have short stature [Stein et al 2004].
- It is unclear from the literature if the use of GH is beneficial in individuals with Costello syndrome with partial GH deficiency. An abnormal GH response on testing and a good initial growth response have been reported [Legault et al 2001].
- Cardiac hypertrophy. Whether the anabolic actions of GH accelerate preexisting cardiac hypertrophy is not known, but early descriptive studies do not suggest a clear association [Lin et al 2002, Lin et al 2011]. In rare cases, cardiomyopathy has progressed after initiation of GH treatment; whether the relationship was causal or coincidental is unknown (see, e.g., Kerr et al [2003]).
- Malignancy. The effect of GH on tumor predisposition has not been determined. Two reports have raised the possibility of an association.
- Bladder carcinoma occurred in an individual age 16 years treated with GH [Gripp et al 2000].
- A rhabdomyosarcoma was diagnosed in an individual age 26 months receiving GH from age 12 months [Kerr et al 2003].
- On review of 35 affected individuals, 16 had documented GH deficiency (46%). Thirteen of these 16 received GH treatment (37%). In this cohort, 7/35 had tumors, including four rhabdomyosarcomas, one pituitary adenoma, one benign bladder tumor, and one bladder carcinoma. Of the seven with tumors, four (57%) were naïve to GH at the time of tumor diagnosis and three (43%) received GH prior to tumor diagnosis. Of the individuals who developed rhabdomyosarcoma, two never received GH, one received GH after tumor diagnosis, and one received GH prior to the diagnosis of rhabdomyosarcoma [Rauen et al 2008].
Developmental Delay / Intellectual Disability Management Issues
The following information represents typical management recommendations for individuals with developmental delay / intellectual disability in the United States; standard recommendations may vary from country to country.
Ages 0-3 years. Referral to an early intervention program is recommended for access to occupational, physical, speech, and feeding therapy as well as infant mental health services, special educators, and sensory impairment specialists. In the US, early intervention is a federally funded program available in all states that provides in-home services to target individual therapy needs.
Ages 3-5 years. In the US, developmental preschool through the local public school district is recommended. Before placement, an evaluation is made to determine needed services and therapies and an individualized education plan (IEP) is developed for those who qualify based on established motor, language, social, or cognitive delay. The early intervention program typically assists with this transition. Developmental preschool is center based; for children too medically unstable to attend, home-based services are provided.
All ages. Consultation with a developmental pediatrician is recommended to ensure the involvement of appropriate community, state, and educational agencies (US) and to support parents in maximizing quality of life. Some issues to consider:
- IEP services:
- An IEP provides specially designed instruction and related services to children who qualify.
- IEP services will be reviewed annually to determine whether any changes are needed.
- Special education law requires that children participating in an IEP be in the least restrictive environment feasible at school and included in general education as much as possible, when and where appropriate.
- Vision consultants should be a part of the child's IEP team to support access to academic material.
- PT, OT, and speech services will be provided in the IEP to the extent that the need affects the child's access to academic material. Beyond that, private supportive therapies based on the affected individual's needs may be considered. Specific recommendations regarding type of therapy can be made by a developmental pediatrician.
- As a child enters the teen years, a transition plan should be discussed and incorporated in the IEP. For those receiving IEP services, the public school district is required to provide services until age 21.
- A 504 plan (Section 504: a US federal statute that prohibits discrimination based on disability) can be considered for those who require accommodations or modifications such as front-of-class seating, assistive technology devices, classroom scribes, extra time between classes, modified assignments, and enlarged text.
- Developmental Disabilities Administration (DDA) enrollment is recommended. DDA is a US public agency that provides services and support to qualified individuals. Eligibility differs by state but is typically determined by diagnosis and/or associated cognitive/adaptive disabilities.
- Families with limited income and resources may also qualify for supplemental security income (SSI) for their child with a disability.
Motor Dysfunction
Gross motor dysfunction
- Physical therapy is recommended to maximize mobility and to reduce the risk for later-onset orthopedic complications (e.g., contractures, scoliosis, hip dislocation, osteopenia).
- Consider use of durable medical equipment and positioning devices as needed (e.g., wheelchairs, walkers, bath chairs, orthotics, adaptive strollers).
Fine motor dysfunction. Occupational therapy is recommended for difficulty with fine motor skills that affect adaptive function such as feeding, grooming, dressing, and writing.
Oral motor dysfunction should be assessed at each visit and clinical feeding evaluations may be considered. When feeding dysfunction is severe, which is typical in infants and children, an NG-tube or G-tube may be necessary. G-tube placement should be considered sooner than in typical individuals as the prolonged oral feeding difficulties are to be expected.
Communication issues. Consider evaluation for alternative means of communication (e.g., augmentative and alternative communication [AAC]) for individuals who have expressive language difficulties. An AAC evaluation can be completed by a speech-language pathologist who has expertise in the area. The evaluation will consider cognitive abilities and sensory impairments to determine the most appropriate form of communication. AAC devices can range from low-tech, such as picture exchange communication, to high-tech, such as voice-generating devices. Contrary to popular belief, AAC devices do not hinder verbal development of speech, but rather support optimal speech and language development.
Neurobehavioral/Psychiatric Concerns
Children may qualify for and benefit from interventions used in treatment of autism spectrum disorder, including applied behavior analysis (ABA). ABA therapy is targeted to the individual child's behavioral, social, and adaptive strengths and weaknesses and typically performed one on one with a board-certified behavior analyst.
Consultation with a developmental pediatrician may be helpful in guiding parents through appropriate behavior management strategies or providing prescription medications, such as medication used to treat attention-deficit/hyperactivity disorder, when necessary.
Concerns about serious aggressive or destructive behavior can be addressed by a pediatric psychiatrist.
Surveillance
To monitor existing manifestations, the individual's response to supportive care, and the emergence of new manifestations, the evaluations summarized in Table 7 are recommended.
Table 7.
Costello Syndrome: Recommended Surveillance
Agents/Circumstances to Avoid
Aggressive feeding therapy in infancy and early childhood is not likely to improve oral intake and may result in oral aversion.
Neuroblastoma screening by measuring catecholamine metabolites is not helpful because elevated values were observed in individuals without an identifiable tumor.
Evaluation of Relatives at Risk
See Genetic Counseling for issues related to testing of at-risk relatives for genetic counseling purposes.
Therapies Under Investigation
Search ClinicalTrials.gov in the US and EU Clinical Trials Register in Europe for information on clinical studies for a wide range of diseases and conditions.
Genetic Counseling
Genetic counseling is the process of providing individuals and families with information on the nature, mode(s) of inheritance, and implications of genetic disorders to help them make informed medical and personal decisions. The following section deals with genetic risk assessment and the use of family history and genetic testing to clarify genetic status for family members; it is not meant to address all personal, cultural, or ethical issues that may arise or to substitute for consultation with a genetics professional. —ED.
Mode of Inheritance
HRAS-related Costello syndrome (Costello syndrome) is an autosomal dominant disorder typically caused by a de novo pathogenic variant.
Risk to Family Members
Parents of a proband
- To date, most individuals with Costello syndrome have the disorder as the result of a de novo HRAS pathogenic variant.An association with advanced parental age has been documented [Giannoulatou et al 2013]. Most but not all de novo pathogenic variants arise in the paternal germline; Sol-Church et al [2006] reported 14 de novo pathogenic variants of paternal origin and two of maternal origin.
- Rarely, an individual with Costello syndrome has the disorder as the result of an HRAS pathogenic variant inherited from a heterozygous parent. Vertical transmission has been reported in two families with the rare, attenuated phenotype of Costello syndrome (see Genotype-Phenotype Correlations) [Gripp et al 2012, Gripp et al 2015].
- Molecular genetic testing is recommended for the parents of the proband to evaluate their genetic status and inform recurrence risk assessment.
- If the pathogenic variant identified in the proband is not identified in either parent and parental identity testing has confirmed biological maternity and paternity, the following possibilities should be considered:
- The proband has a de novo pathogenic variant.
- The proband inherited a pathogenic variant from a parent with germline (or somatic and germline) mosaicism.* Note: Testing of parental leukocyte DNA may not detect all instances of somatic mosaicism and will not detect a pathogenic variant that is present in the germ (gonadal) cells only.* A parent with somatic and germline mosaicism for an HRAS pathogenic variant may show patchy skin findings only (as reported in the mosaic father of an individual with typical Costello syndrome [Sol-Church et al 2009]).
Sibs of a proband. The risk to the sibs of the proband depends on the genetic status of the proband's parents:
- If the HRAS pathogenic variant identified in the proband cannot be detected in the leukocyte DNA of either parent, the recurrence risk to sibs is slightly greater than that of the general population because of the possibility of parental germline mosaicism. Recurrence of Costello syndrome in sibs has been reported and is suspected to be the result of germline mosaicism in a parent [Zampino et al 1993, Johnson et al 1998, Gripp et al 2011b].
- If a parent of the proband has the pathogenic variant identified in the proband, the risk to the sibs of inheriting the pathogenic variant is 50%.
- If the parents have not been tested for the HRAS pathogenic variant but are clinically unaffected, the risk to the sibs of a proband appears to be low. However, sibs of a proband with clinically unaffected parents are still presumed to be at increased risk for Costello syndrome because of the possibility of parental mosaicism.
Offspring of a proband
- Each child of an individual with an attenuated phenotype has a 50% chance of inheriting the HRAS pathogenic variant.
- Individuals with characteristic Costello syndrome typically do not reproduce.
Other family members. If the proband has Costello syndrome as the result of a de novo pathogenic variant, other family members are not at increased risk.
Related Genetic Counseling Issues
Family planning
- The optimal time for determination of genetic risk and discussion of the availability of prenatal/preimplantation genetic testing is before pregnancy.
- It is appropriate to offer genetic counseling (including discussion of potential risks to offspring and reproductive options) to parents of children with Costello syndrome.
Prenatal Testing and Preimplantation Genetic Testing
Molecular genetic testing. If an HRAS pathogenic variant has been identified in an affected family member, prenatal and preimplantation genetic testing are possible.
Ultrasound examination in a pregnancy not known to be at increased risk for Costello syndrome. The fetal phenotype of Costello syndrome (including increased nuchal thickness, macrocephaly, mild shortness of the long bones, polyhydramnios, and fetal tachycardia) is not unique, and as a rare disorder, Costello syndrome is often not considered. However, the presence of severe polyhydramnios in the pregnancy of a fetus with normal chromosome analysis or chromosome microarray and fetal atrial tachycardia may warrant consideration of the diagnosis of Costello syndrome.
Differences in perspective may exist among medical professionals and within families regarding the use of prenatal testing. While most centers would consider use of prenatal testing to be a personal decision, discussion of these issues may be helpful.
Resources
GeneReviews staff has selected the following disease-specific and/or umbrella support organizations and/or registries for the benefit of individuals with this disorder and their families. GeneReviews is not responsible for the information provided by other organizations. For information on selection criteria, click here.
- Costello Syndrome Family NetworkEmail: [email protected]
- CostelloKids UKUnited Kingdom
- MedlinePlus
- RASopathies NetworkEmail: [email protected]
- Children's Craniofacial AssociationPhone: 800-535-3643Email: [email protected]
- MAGIC FoundationPhone: 630-836-8200Email: [email protected]
Molecular Genetics
Information in the Molecular Genetics and OMIM tables may differ from that elsewhere in the GeneReview: tables may contain more recent information. —ED.
Table A.
HRAS-Related Costello Syndrome: Genes and Databases
Table B.
OMIM Entries for HRAS-Related Costello Syndrome (View All in OMIM)
Molecular Pathogenesis
The RAS oncogenes, HRAS, KRAS, and NRAS, encode 21-kd proteins collectively called p21RAS. The p21RAS proteins are localized to the inner plasma membrane, where they bind GDP and GTP and have low intrinsic GTPase activity [Corbett & Alber 2001]. The GDP-bound conformation is the inactive state of the RAS molecule. An extracellular stimulus – for example, through growth factor receptors – initiates release of GDP and subsequent binding of GTP. The GTP-bound form is active and permits signal transduction. This transmission of mitogenic and growth signals allows the widely expressed RAS proteins to regulate cell proliferation, differentiation, transformation, and apoptosis. Hydrolysis of the bound GTP to GDP reverses the active state. The low intrinsic GTPase activity of RAS proteins is increased through GTPase-activating proteins (GAPs) and other regulators including neurofibromin protein (see Neurofibromatosis Type 1). Normally, most p21RAS within a cell is present in an inactive GDP-bound state.
HRAS is an oncogene that is aberrantly activated in sporadic tumors. Therefore, much of what is known about the abnormal gene product has been learned through cancer research because the germline HRAS single-nucleotide pathogenic variants that cause Costello syndrome are identical to the somatic HRAS single-nucleotide pathogenic variants observed in sporadic malignant tumors unrelated to Costello syndrome. Activating single-nucleotide pathogenic variants leading to an amino acid substitution at positions 12, 13, and 61 are the most common in malignant tumors; less commonly, amino acids 59, 63, 116, 117, 119, or 146 are affected. These pathogenic missense variants result in constitutive activation of the abnormal protein product, and thus lead to increased signaling through the Ras-MAPK [Sol-Church & Gripp 2009] and the PI3K-AKT-MTOR pathways [Rosenberger et al 2009]. A more complex dysregulation of the signaling pathways was reported for the rare HRAS p.Gly60Asp pathogenic variant [Gripp et al 2015].
Amino acid changes lead either to decreased GTPase activity (if amino acids 12, 13, 59, 61, or 63 are involved), so that oncogenic RAS mutated proteins are locked in the active GTP-bound state, or decreased nucleotide affinity and, hence, increased exchange of bound GDP for cytosolic GTP (if amino acids 116, 117, 119, or 146 are affected). Single-nucleotide pathogenic variants cause an accumulation of activated RAS-GTP complexes, leading to continuous signal transduction by facilitating accumulation of constitutively active, GTP-bound RAS protein.
Kerr et al [2003] identified loss of heterozygosity for 11p15.5 in rhabdomyosarcoma from individuals with Costello syndrome, suggesting that loss of the wild type allele is the second hit in tumor development. This theory is supported by the loss of the wild type allele in a rhabdomyosarcoma demonstrated by Estep et al [2006] and the monoallelic expression in a tumor, but not in fibroblasts, reported by Aoki et al [2005]. Robbins et al [2016] studied 11 rhabdomyosarcoma samples from eight unrelated individuals with Costello syndrome. Eight tumors showed complete paternal uniparental disomy (UPD) of chromosome 11 (pUPD11), whereas two had UPD only at 11p. A second primary rhabdomyosarcoma showed UPD limited to 11p15.5 [Robbins et al 2016].
Mechanism of disease causation. Gain of function
Table 8.
HRAS Pathogenic Variants Referenced in This GeneReview
Chapter Notes
Author Notes
Dr Karen Gripp is a codirector of the Professional Advisory Committee of the Costello Syndrome Family Network. She is a member of the ClinGen expert panels on RASopathies and inherited cancer syndromes.
Dr Nicole Weaver is a member of the Professional Advisory Committee of the Costello Syndrome Family Network. She is a member of the ClinGen expert panel on congenital heart disease.
Drs Gripp and Weaver are actively involved in clinical research regarding individuals with Costello syndrome. They would be happy to communicate with persons who have any questions regarding diagnosis of Costello syndrome or other considerations.
Acknowledgments
Special thanks to Sandra Taylor, Executive Director, and Mary Ernst, President of the Costello Syndrome Family Network; Lisa Schoyer, Chair of the Research Advisory Committee, and Colin Stone, President of the International Costello Syndrome Support Group; the individuals with Costello syndrome and their families; and our colleagues on the Professional Advisory Committee.
Author History
Karen W Gripp, MD, FAAP, FACMG (2006-present)
Angela E Lin, MD, FAAP, FACMG; Harvard Medical School (2006-2019)
Katherine A Rauen, MD, PhD; University of California, Davis (2019-2023)
K Nicole Weaver, MD, FAAP, FACMG (2023-present)
Revision History
- 21 December 2023 (gm) Comprehensive update posted live
- 29 August 2019 (bp) Comprehensive update posted live
- 12 January 2012 (me) Comprehensive update posted live
- 19 May 2009 (me) Comprehensive update posted live
- 29 August 2006 (me) Review posted live
- 2 March 2006 (kg) Original submission
References
Literature Cited
- Abe Y, Aoki Y, Kuriyama S, Kawame H, Okamoto N, Kurasawa K, Ohashi H, Mizuno S, Ogata T, Kure S, Niihori T, Matsubara Y. Epidemiological features of Costello syndrome and Cardio-facio-cutaneous syndrome: findings from the first nationwide survey. Chicago, IL: International Meeting on Genetic Syndromes of the Ras/MAPK Pathway; 2011. [PubMed: 22495831]
- Alexander S, Ramadan D, Alkhayyat H, Al-Sharkawi I, Backer KC, El-Sabban F, Hussain K. Costello syndrome and hyperinsulinemic hypoglycemia. Am J Med Genet. 2005;139:227–30. [PubMed: 16278907]
- Andelfinger G, Marquis C, Raboisson MJ, Théoret Y, Waldmüller S, Wiegand G, Gelb BD, Zenker M, Delrue MA, Hofbeck M. Hypertrophic Cardiomyopathy in Noonan Syndrome Treated by MEK-Inhibition. J Am Coll Cardiol. 2019;73:2237-9. [PMC free article: PMC6916648] [PubMed: 31047013]
- Aoki Y, Niihori T, Kawame H, Kurosawa K, Ohashi H, Tanaka Y, Filocamo M, Kato K, Suzuki Y, Kure S, Matsubara Y. Germline mutations in HRAS proto-oncogene cause Costello syndrome. Nat Genet. 2005;37:1038–40. [PubMed: 16170316]
- Astiazaran-Symonds E, Ney GM, Higgs C, Oba L, Srivastava R, Livinski AA, Rosenberg PS, Stewart DR. Cancer in Costello syndrome: a systematic review and meta-analysis. Br J Cancer. 2023;128:2089-96. doi: .10.1038/s41416-023-02229-7 [PMC free article: PMC10205753] [PubMed: 36966234] [CrossRef]
- Axelrad ME, Glidden R, Nicholson L, Gripp KW. Adaptive skills, cognitive, and behavioral characteristics of Costello syndrome. Am J Med Genet A. 2004;128A:396–400. [PubMed: 15264285]
- Axelrad ME, Nicholson L, Stabley DL, Sol-Church K, Gripp KW. Longitudinal assessment of cognitive characteristics in Costello syndrome. Am J Med Genet A. 2007;143A:3185–93. [PubMed: 17963256]
- Axelrad ME, Schwartz DD, Fehlis JE, Hopkins E, Stabley DL, Sol-Church K, Gripp KW. Longitudinal course of cognitive, adaptive, and behavioral characteristics in Costello syndrome. Am J Med Genet A. 2009;149A:2666–72. [PMC free article: PMC2787985] [PubMed: 19919001]
- Axelrad ME, Schwartz DD, Katzenstein JM, Hopkins E, Gripp KW. Neurocognitive, adaptive, and behavioral functioning of individuals with Costello syndrome: a review. Am J Med Genet C Semin Med Genet. 2011;157C:115–22. [PubMed: 21495179]
- Bertola D, Buscarilli M, Stabley DL, Baker L, Doyle D, Bartholomew DW, Sol-Church K, Gripp KW. Phenotypic spectrum of Costello syndrome individuals harboring the rare HRAS mutation p.Gly13Asp. Am J Med Genet. 2017;173:1309–18. [PMC free article: PMC5397353] [PubMed: 28371260]
- Borochowitz Z, Pavone L, Mazor G, Rizzo R, Dar H. New multiple congenital anomalies: mental retardation syndrome (MCA/MR) with facio-cutaneous-skeletal involvement. Am J Med Genet. 1992;43:678–85. [PubMed: 1621757]
- Calandrelli R, D'Apolito G, Marco P, Zampino G, Tartaglione T, Colosimo C. Costello syndrome: Analysis of the posterior cranial fossa in children with posterior fossa crowding. Neuroradiol J. 2015;28:254–8. [PMC free article: PMC4757298] [PubMed: 26246091]
- Corbett KD, Alber T. The many faces of Ras: recognition of small GTP-binding proteins. Trends Biochem Sci. 2001;26:710–6. [PubMed: 11738594]
- Costello JM. A new syndrome. NZ Med J. 1971;74:397.
- Costello JM. A new syndrome: mental subnormality and nasal papillomata. Aust Paediatr J. 1977;13:114–8. [PubMed: 907573]
- Costello JM. Costello syndrome: update on the original cases and commentary. Am J Med Genet. 1996;62:199–201. [PubMed: 8882404]
- Della Marca G, Vasta I, Scarano E, Rigante M, De Feo E, Mariotti P, Rubino M, Vollono C, Mennuni GF, Tonali P, Zampino G. Obstructive sleep apnea in Costello syndrome. Am J Med Genet A. 2006;140:257–62. [PubMed: 16419102]
- Delrue MA, Chateil JF, Arveiler B, Lacombe D. Costello syndrome and neurological abnormalities. Am J Med Genet A. 2003;123A:301–5. [PubMed: 14608654]
- Der Kaloustian VM, Moroz B, McIntosh N, Watters AK, Blaichman S. Costello syndrome. Am J Med Genet. 1991;41:69–73. [PubMed: 1951465]
- Detweiler S, Thacker MM, Hopkins E, Conway L, Gripp KW. Orthopedic manifestations and implications for individuals with Costello syndrome. Am J Med Genet. 2013;161A:1940–9. [PubMed: 23813656]
- Estep AL, Tidyman WE, Teitell MA, Cotter PD, Rauen KA. HRAS mutations in Costello syndrome: detection of constitutional activating mutations in codon 12 and 13 and loss of wild-type allele in malignancy. Am J Med Genet A. 2006;140:8–16. [PubMed: 16372351]
- Geddes GC, Parent JJ, Lander J, Jeewa A, Ware SM, Villa C, Chatfield KC, Weaver KN. MEK Inhibition Improves Cardiomyopathy in Costello Syndrome. J Am Coll Cardiol. 2023;81:1439-41. [PubMed: 37019585]
- Giannoulatou E, McVean G, Taylor IB, McGowan SJ, Maher GJ, Iqbal Z, Pfeifer SP, Turner I, Burkitt Wright EM, Shorto J, Itani A, Turner K, Gregory L, Buck D, Rajpert-De Meyts E, Looijenga LH, Kerr B, Wilkie AO, Goriely A. Contributions of intrinsic mutation rate and selfish selection to levels of de novo HRAS mutations in the paternal germline. Proc Natl Acad Sci USA. 2013;110:20152–7. [PMC free article: PMC3864328] [PubMed: 24259709]
- Girisha KM, Lewis LE, Phadke SR, Kutsche K. Costello syndrome with severe cutis laxa and mosaic HRAS G12S mutation. Am J Med Genet A. 2010;152A:2861–4. [PubMed: 20979192]
- Gomez-Ospina N, Kuo C, Ananth AL, Myers A, Brennan ML, Stevenson DA, Bernstein JA, Hudgins L. Respiratory system involvement in Costello syndrome. Am J Med Genet. 2016;170:1849–57. [PMC free article: PMC5509842] [PubMed: 27102959]
- Goodwin AF, Oberoi S, Landan M, Charles C, Massie JC, Fairley C, Rauen KA, Klein OD. Craniofacial and dental development in Costello syndrome. Am J Med Genet. 2014;164A:1425–30. [PMC free article: PMC4115793] [PubMed: 24668879]
- Grant AR, Cushman BJ, Cave H, Dillon MW, Gelb BD, Gripp KW, Lee JA, Mason-Suares H, Rauen KA, Tartaglia M, Vincent LM, Zenker M. Assessing the gene-disease association of 19 genes with the RASopathies using the ClinGen gene curation framework. Hum Mutat. 2018;39:1485–93. [PMC free article: PMC6326381] [PubMed: 30311384]
- Gremer L, De Luca A, Merbitz-Zahradnik T, Dallapiccola B, Morlot S, Tartaglia M, Kutsche K, Ahmadian MR, Rosenberger G. Duplication of Glu37 in the switch I region of HRAS impairs effector/GAP binding and underlies Costello syndrome by promoting enhanced growth factor-dependent MAPK and AKT activation. Hum Mol Genet. 2010;19:790–802. [PubMed: 19995790]
- Gripp KW. Tumor predisposition in Costello syndrome. Am J Med Genet C Semin Med Genet. 2005;137C:72–7. [PubMed: 16010679]
- Gripp KW, Baker L, Robbins KM, Stabley DL, Bellus GA, Kolbe V, Nauth T, Rosenberger G. The novel duplication HRAS c.186_206dup p.(Glu62_Arg68dup): clinical and functional aspects. Eur J Hum Genet. 2020;28:1548-54. [PMC free article: PMC7576819] [PubMed: 32499600]
- Gripp KW, Demmer LA. Keratoconus in Costello syndrome. Am J Med Genet. 2013;161A:1132–6. [PubMed: 23494969]
- Gripp KW, Hopkins E, Doyle D, Dobyns WB. High incidence of progressive postnatal cerebellar enlargement in Costello syndrome: brain overgrowth associated with HRAS mutations as the likely cause of structural brain and spinal cord abnormalities. Am J Med Genet A. 2010;152A:1161–8. [PMC free article: PMC4910816] [PubMed: 20425820]
- Gripp KW, Hopkins E, Serrano A, Leonard N J, Stabley DL, Sol-Church K. Transmission of the rare HRAS mutation (c. 173C>T; p.T58I) further illustrates its attenuated phenotype. Am J Med Genet Part A. 2012;158A:1095–101. [PMC free article: PMC4164267] [PubMed: 22488832]
- Gripp KW, Hopkins E, Sol-Church K, Stabley DL, Axelrad ME, Doyle D, Dobyns WB, Hudson C, Johnson J, Tenconi R, Graham GE, Sousa AB, Heller R, Piccione M, Corsello G, Herman GE, Tartaglia M, Lin AE. Phenotypic analysis of individuals with Costello syndrome due to HRAS p.G13C. Am J Med Genet A. 2011a;155A:706–16. [PMC free article: PMC4166651] [PubMed: 21438134]
- Gripp KW, Innes AM, Axelrad ME, Gillan TL, Parboosingh JS, Davies C, Leonard NJ, Lapointe M, Doyle D, Catalano S, Nicholson L, Stabley DL, Sol-Church K. Costello syndrome associated with novel germline HRAS mutations: an attenuated phenotype? Am J Med Genet A. 2008;146A:683–90. [PubMed: 18247425]
- Gripp KW, Kawame H, Viskochil DH, Nicholson L. Elevated catecholamine metabolites in patients with Costello syndrome. Am J Med Genet A. 2004;128A:48–51. [PubMed: 15211656]
- Gripp KW, Kolbe V, Brandenstein LI, Rosenberger G. Attenuated phenotype of Costello syndrome and early death in a patient with an HRAS Mutation (c.179G>T; p.Gly60Val) affecting signalling dynamics. Clin Genet. 2017;92:332–7. [PubMed: 28139825]
- Gripp KW, Lin AE. Costello syndrome: A Ras/mitogen activated protein kinase pathway syndrome (rasopathy) resulting from HRAS germline mutations. Genet Med. 2012;14:285–92. [PubMed: 22261753]
- Gripp KW, Lin AE, Nicholson L, Allen W, Cramer A, Jones KL, Kutz W, Peck D, Rebolledo MA, Wheeler PG, Wilson W, Al-Rahawan MM, Stabley DL, Sol-Church K. Further delineation of the phenotype resulting from BRAF or MEK1 germline mutations helps differentiate cardio-facio-cutaneous syndrome from Costello syndrome. Am J Med Genet A. 2007;143A:1472–80. [PubMed: 17551924]
- Gripp KW, Lin AE, Stabley DL, Nicholson L, Scott CI Jr, Doyle D, Aoki Y, Matsubara Y, Zackai EH, Lapunzina P, Gonzalez-Meneses A, Holbrook J, Agresta CA, Gonzalez IL, Sol-Church K. HRAS mutation analysis in Costello syndrome: genotype and phenotype correlation. Am J Med Genet A. 2006a;140:1–7. [PubMed: 16329078]
- Gripp KW, Morse LA, Axelrad M, Chatfield KC, Chidekel A, Dobyns W, Doyle D, Kerr B, Lin AE, Schwartz DD, Sibbles BJ, Siegel D, Shankar SP, Stevenson DA, Thacker MM, Weaver KN, White SM, Rauen KA. Costello syndrome: clinical phenotype, genotype, and management guidelines. Am J Med Genet. 2019;179:1725–44. [PMC free article: PMC8238015] [PubMed: 31222966]
- Gripp KW, Robbins KM, Sheffield BS, Lee AF, Patel MS, Yip S, Doyle D, Stabley D, Sol-Church K. Paternal uniparental disomy 11p15.5 in the pancreatic nodule of an infant with Costello syndrome: Shared mechanism for hyperinsulinemic hypoglycemia in neonates with Costello and Beckwith-Wiedemann syndrome and somatic loss of heterozygosity in Costello syndrome driving clonal expansion. Am J Med Genet. 2016;170:559–64. [PMC free article: PMC4784973] [PubMed: 26572961]
- Gripp KW, Scott CI Jr, Nicholson L, Figueroa TE. Second case of bladder carcinoma in a patient with Costello syndrome. Am J Med Genet. 2000;90:256–9. [PubMed: 10678668]
- Gripp KW, Scott CI Jr, Nicholson L, McDonald-McGinn DM, Ozeran JD, Jones MC, Lin AE, Zackai EH. Five additional Costello syndrome patients with rhabdomyosarcoma: proposal for a tumor screening protocol. Am J Med Genet. 2002;108:80–7. [PubMed: 11857556]
- Gripp KW, Sol-Church K, Smpokou P, Graham GE, Stevenson DA, Hanson H, Viskochil DH, Baker LC, Russo B, Gardner N, Stabley DL, Kolbe V, Rosenberger G. An attenuated phenotype of Costello syndrome in three unrelated individuals with a HRAS c.179G>A (p.Gly60Asp) mutation with uncommon functional consequences. Am J Med Genet A. 2015;167A:2085–97. [PMC free article: PMC4830354] [PubMed: 25914166]
- Gripp KW, Stabley DL, Geller PL, Hopkins E, Stevenson DA, Carey JC, Sol-Church K. Molecular confirmation of HRAS p.G12S in siblings with Costello syndrome. Am J Med Genet A. 2011b;155A:2263–8. [PMC free article: PMC3158836] [PubMed: 21834037]
- Gripp KW, Stabley DL, Nicholson L, Hoffman JD, Sol-Church K. Somatic mosaicism for an HRAS mutation causes Costello syndrome. Am J Med Genet A. 2006b;140:2163–9. [PubMed: 16969868]
- Groesser L, Herschberger E, Ruetten A, Ruivenkamp C, Lopriore E, Zutt M, Langmann T, Singer S, Klingseisen L, Schneider-Brachert W, Toll A, Real FX, Landthaler M, Hafner C. Postzygotic HRAS and KRAS mutations cause nevus sebaceous and Schimmelpenning syndrome. Nat Genet. 2012;44:783-7. [PubMed: 22683711]
- Hall BD, Berbereich FR, Berbereich MS. AMICABLE syndrome: A new unique disorder involving facial, cardiac, ectodermal, growth and intellectual abnormalities. Proc Greenwood Genet Center. 1990;9:103A.
- Hennekam RC. Costello syndrome: an overview. Am J Med Genet Part C Semin Med Genet. 2003;117C:42–8. [PubMed: 12561057]
- Hopkins E, Lin AE, Krepkovich KE, Axelrad ME, Sol-Church K, Stabley DL, Hossain J, Gripp KW. Living with Costello syndrome: Quality of life issues in older individuals. Am J Med Genet A. 2010;152A:84–90. [PubMed: 20034064]
- Johnson B, Goldberg-Strassler D, Gripp K, Thacker M, Leoni C, Stevenson D. Function and disability in children with Costello syndrome and cardiofaciocutaneous syndrome. Am J Med Genet. 2015;167A:40–4. [PubMed: 25346259]
- Johnson JP, Golabi M, Norton ME, Rosenblatt RM, Feldman GM, Yang SP, Hall BD, Fries MH, Carey JC. Costello syndrome: phenotype, natural history, differential diagnosis, and possible cause. J Pediatr. 1998;133:441–8. [PubMed: 9738731]
- Kawame H, Matsui M, Kurosawa K, Matsuo M, Masuno M, Ohashi H, Fueki N, Aoyama K, Miyatsuka Y, Suzuki K, Akatsuka A, Ochiai Y, Fukushima Y. Further delineation of the behavioral and neurologic features in Costello syndrome. Am J Med Genet A. 2003;118A:8–14. [PubMed: 12605434]
- Kerr B, Allanson J, Delrue MA, Gripp KW, Lacombe D, Lin AE, Rauen KA. The diagnosis of Costello syndrome: nomenclature in Ras/MAPK pathway disorders. Am J Med Genet A. 2008;146A:1218–20. [PubMed: 18386799]
- Kerr B, Delrue MA, Sigaudy S, Perveen R, Marche M, Burgelin I, Stef M, Tang B, Eden OB, O'Sullivan J, De Sandre-Giovannoli A, Reardon W, Brewer C, Bennett C, Quarell O, M'Cann E, Donnai D, Stewart F, Hennekam R, Cavé H, Verloes A, Philip N, Lacombe D, Levy N, Arveiler B, Black G. Genotype-phenotype correlation in Costello syndrome: HRAS mutation analysis in 43 cases. J Med Genet. 2006;43:401–5. [PMC free article: PMC2564514] [PubMed: 16443854]
- Kerr B, Einaudi MA, Clayton P, Gladman G, Eden T, Saunier P, Genevieve D, Philip N. Is growth hormone treatment beneficial or harmful in Costello syndrome? J Med Genet. 2003;40:e74. [PMC free article: PMC1735496] [PubMed: 12807973]
- Kratz CP, Rapisuwon S, Reed H, Hasle H, Rosenberg PS. Cancer in Noonan, Costello, cardiofaciocutaneous and LEOPARD syndromes. Am J Med Genet C Semin Med Genet. 2011;157C:83–9. [PMC free article: PMC3086183] [PubMed: 21500339]
- Leegaard A, Gregersen PA, Nielsen TØ, Bjerre JV, Handrup MM. Succesful MEK-inhibition of severe hypertrophic cardiomyopathy in RIT1-related Noonan Syndrome. Eur J Med Genet. 2022;65:104630. [PubMed: 36184070]
- Legault L, Gagnon C, Lapointe N. Growth hormone deficiency in Costello syndrome: a possible explanation for the short stature. J Pediatr. 2001;138:151–2. [PubMed: 11148542]
- Leoni C, Paradiso FV, Foschi N, Tedesco M, Pierconti F, Silvaroli S, Diego MD, Birritella L, Pantaleoni F, Rendeli C, Onesimo R, Viscogliosi G, Bassi P, Nanni L, Genuardi M, Tartaglia M, Zampino G. Prevalence of bladder cancer in Costello syndrome: New insights to drive clinical decision-making. Clin Genet. 2022;101:454-8. [PubMed: 35038173]
- Leoni C, Romeo DM, Pelliccioni M, Di Già M, Onesimo R, Giorgio V, Flex E, Tedesco M, Tartaglia M, Rigante D, Valassina A, Zampino G. Musculo-skeletal phenotype of Costello syndrome and cardio-facio-cutaneous syndrome: insights on the functional assessment status. Orphanet J Rare Dis. 2021;16:43. [PMC free article: PMC7821553] [PubMed: 33482860]
- Leoni C, Stevenson DA, Martini L, De Sanctis R, Mascolo G, Pantaleoni F, De Santis S, La Torraca I, Persichilli S, Caradonna P, Tartaglia M, Zampino G. Decreased bone mineral density in Costello syndrome. Mol Genet Metab. 2014;111:41–5. [PubMed: 24246682]
- Levin MD, Saitta SC, Gripp KW, Wenger TL, Ganesh J, Kalish JM, Epstein MR, Smith R, Czosek RJ, Ware SM, Goldenberg P, Myers A, Chatfield KC, Gillespie MJ, Zackai EH, Lin AE. Nonreentrant atrial tachycardia occurs independently of hypertrophic cardiomyopathy in RASopathy patients. Am J Med Genet. 2018;176:1711–22. [PMC free article: PMC6107379] [PubMed: 30055033]
- Lim YH, Ovejero D, Derrick KM; Yale Center for Mendelian Genomics; Collins MT, Choate KA. Cutaneous skeletal hypophosphatemia syndrome (CSHS) is a multilineage somatic mosaic RASopathy. J Am Acad Dermatol. 2016;75:420-7. [PMC free article: PMC5004488] [PubMed: 27444071]
- Lin AE, Alexander ME, Colan SD, Kerr B, Rauen KA, Noonan J, Baffa J, Hopkins E, Sol-Church K, Limongelli G, Digilio MC, Marino B, Ines AM, Aoki Y, Silberbach M, Del-Rue MA, While SM, Hamilton RM, O’Connor W, Grossfeld PD, Smoot LB, Padera RF, Gripp KW. Clinical, pathological and molecular analyses of cardiovascular abnormalities in Costello syndrome: A Ras/MAPK Pathway syndrome. Am J Med Genet A. 2011;155A:486–507. [PubMed: 21344638]
- Lin AE, Grossfeld PD, Hamilton RM, Smoot L, Gripp KW, Proud V, Weksberg R, Wheeler P, Picker J, Irons M, Zackai E, Marino B, Scott CI Jr, Nicholson L. Further delineation of cardiac abnormalities in Costello syndrome. Am J Med Genet. 2002;111:115–29. [PubMed: 12210337]
- Lo FS, Lin JL, Kuo MT, Chiu PC, Shu SG, Chao MC, Lee YJ, Lin SP. Noonan syndrome caused by germline KRAS mutation in Taiwan: report of two patients and a review of the literature. Eur J Pediatr. 2009;168:919–23. [PubMed: 18958496]
- Lo IF, Brewer C, Shannon N, Shorto J, Tang B, Black G, Soo MT, Ng DK, Lam ST, Kerr B. Severe neonatal manifestations of Costello syndrome. J Med Genet. 2008;45:167–71. [PubMed: 18039947]
- Lorenz S, Petersen C, Kordaß U, Seidel H, Zenker M, Kutsche K. Two cases with severe lethal course of Costello syndrome associated with HRAS p.G12C and p.G12D. Eur J Med Genet. 2012;55:615-9. [PubMed: 22926243]
- Marukian NV, Levinsohn JL, Craiglow BG, Milstone LM, Choate KA. Palmoplantar Keratoderma in Costello Syndrome Responsive to Acitretin. Pediatr Dermatol. 2017;34:160–162. [PubMed: 28008647]
- McCormick EM, Hopkins E, Conway L, Catalano S, Hossain J, Sol-Church K, Stabley DL, Gripp KW. Assessing genotype-phenotype correlation in Costello syndrome using a severity score. Genet Med. 2013;15:554–7. [PubMed: 23429430]
- Myers A, Bernstein JA, Brennan ML, Curry C, Esplin ED, Fisher J, Homeyer M, Manning MA, Muller EA, Niemi AK, Seaver LH, Hintz SR, Hudgins L. Perinatal features of the RASopathies: Noonan syndrome, cardiofaciocutaneous syndrome and Costello syndrome. Am J Med Genet A. 2014;164A:2814-21. [PubMed: 25250515]
- Niihori T, Aoki Y, Narumi Y, Neri G, Cavé H, Verloes A, Okamoto N, Hennekam RC, Gillessen-Kaesbach G, Wieczorek D, Kavamura MI, Kurosawa K, Ohashi H, Wilson L, Heron D, Bonneau D, Corona G, Kaname T, Naritomi K, Baumann C, Matsumoto N, Kato K, Kure S, Matsubara Y. Germline KRAS and BRAF mutations in cardio-facio-cutaneous syndrome. Nat Genet. 2006;38:294–6. [PubMed: 16474404]
- Ono H, Yamaguchi R, Arai M, Togi S, Ura H, Niida Y, Shimizu A. Schimmelpenning-Feuerstein-Mims syndrome induced by HRAS Gly12Ser somatic mosaic mutation: Case report and literature review. J Dermatol. 2023;50:1213-5. [PubMed: 37170693]
- Paquin A, Hordo C, Kaplan DR, Miller FD. Costello syndrome H-Ras alleles regulate cortical development. Dev Biol. 2009;330:440–51. [PubMed: 19371735]
- Pentiuk S, O'Flaherty T, Santoro K, Willging P, Kaul A. Pureed by gastrostomy tube diet improves gagging and retching in children with fundoplication. JPEN J Parenter Enteral Nutr. 2011;35:375-9. [PubMed: 21527599]
- Pierpont ME, Richards M, Engel WK, Mendelsohn NJ, Summers CG. Retinal dystrophy in two boys with Costello syndrome due to the HRAS p.Gly13Cys mutation. Am J Med Genet. 2017;173:1342–7. [PubMed: 28337834]
- Rauen KA. Distinguishing Costello versus cardio-facio-cutaneous syndrome: BRAF mutations in patients with a Costello phenotype. Am J Med Genet A. 2006;140:1681–3. [PubMed: 16804887]
- Rauen KA, Hefner E, Carrillo K, Taylor J, Messier L, Aoki Y, Gripp KW, Matsubara Y, Proud VK, Hammond P, Allanson JE, Delrue M-A, Axelrad ME, Lin AE, Doyle DA, Kerr B, Carey JC, McCormick F, Silva AJ, Kieran MW, Hinek A, Nguyen TT, Schoyer L. 2008. Molecular aspects, clinical aspects and possible treatment modalities for Costello syndrome: Proceedings from the 1st International Costello Syndrome Research Symposium 2007. Am J Med Genet Part A 146A:1205–17. [PubMed: 18412122]
- Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, Grody WW, Hegde M, Lyon E, Spector E, Voelkerding K, Rehm HL, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17:405–24. [PMC free article: PMC4544753] [PubMed: 25741868]
- Robbins KM, Stabley DL, Holbrook J, Sahraoui R, Sadreameli A, Conard K, Baker L, Gripp KW, Sol-Church K. Paternal uniparental disomy with segmental loss of heterozygosity of chromosome 11 are hallmark characteristics of syndromic and sporadic embryonal rhabdomyosarcoma. Am J Med Genet A. 2016;170:3197-206. [PMC free article: PMC5130350] [PubMed: 27589201]
- Rodriguez-Viciana P, Tetsu O, Tidyman WE, Estep AL, Conger BA, Cruz MS, McCormick F, Rauen KA. Germline mutations in genes within the MAPK pathway cause cardio-facio-cutaneous syndrome. Science. 2006;311:1287–90. [PubMed: 16439621]
- Rosenberger G, Meien S, Kutsche K. Oncogenic HRAS mutations cause prolonged PI3K signaling in response to epidermal growth factor in fibroblasts of patients with Costello syndrome. Hum Mutat. 2009;30:352–62. [PubMed: 19035362]
- Sammon MR, Doyle D, Hopkins E, Sol-Church K, Stabley DL, McGready J, Schulze K, Alade Y, Hoover-Fong J, Gripp KW. Normative growth charts for individuals with Costello syndrome. Am J Med Genet. 2012;158A:2692–9. [PubMed: 22887473]
- Schwartz DD, Katzenstein JM, Highley EJ, Stabley DL, Sol-Church K, Gripp KW, Axelrad ME. Age-related differences in prevalence of autism spectrum disorder symptoms in children and adolescents with Costello syndrome. Am J Med Genet. 2017;173:1294–1300. [PMC free article: PMC5397350] [PubMed: 28374929]
- Schwartz DD, Katzenstein JM, Hopkins E, Stabley DL, Sol-Church K, Gripp KW, Axelrad ME. Verbal memory functioning in adolescents and young adults with Costello syndrome: evidence for relative preservation in recognition memory. Am J Med Genet. 2013;161A:2258–65. [PMC free article: PMC3745536] [PubMed: 23918324]
- Sheffield BS, Yip S, Ruchelli ED, Dunham CP, Sherwin E, Brooks PA, Sur A, Singh A, Human DG, Patel MS, Lee A. Fatal congenital hypertrophic cardiomyopathy and a pancreatic nodule morphologically identical to focal lesion of congenital hyperinsulinism in an infant with Costello syndrome. case report and review of the literature. Pediatr Dev Pathol. 2015;18:237–44. [PubMed: 25668678]
- Shikany AR, Baker L, Stabley DL, Robbins K, Doyle D, Gripp KW, Weaver KN. Medically actionable comorbidities in adults with Costello syndrome. Am J Med Genet A. 2020;182:130-6. [PubMed: 31680412]
- Sol-Church K, Gripp KW. The molecular basis of Costello syndrome. In: Zenker M, ed. Noonan Syndrome and Related Disorders. Monographs in Human Genetics. Basel, Switzerland: Karger. 2009;17:94-103.
- Sol-Church K, Stabley DL, Demmer LA, Agbulos A, Lin AE, Smoot L, Nicholson L, Gripp KW. Male-to-male transmission of Costello syndrome: G12S HRAS germline mutation inherited from a father with somatic mosaicism. Am J Med Genet A. 2009;149A:315–21. [PMC free article: PMC2653086] [PubMed: 19206176]
- Sol-Church K, Stabley DL, Nicholson L, Gonzalez IL, Gripp KW. Paternal bias in parental origin of HRAS mutations in Costello syndrome. Hum Mutat. 2006;27:736–41. [PubMed: 16835863]
- Stein RI, Legault L, Daneman D, Weksberg R, Hamilton J. Growth hormone deficiency in Costello syndrome. Am J Med Genet A. 2004;129A:166–70. [PubMed: 15316968]
- Stenson PD, Mort M, Ball EV, Chapman M, Evans K, Azevedo L, Hayden M, Heywood S, Millar DS, Phillips AD, Cooper DN. The Human Gene Mutation Database (HGMD®): optimizing its use in a clinical diagnostic or research setting. Hum Genet. 2020;139:1197-207. [PMC free article: PMC7497289] [PubMed: 32596782]
- Tidyman WE, Lee HS, Rauen KA. Skeletal muscle pathology in Costello and cardio-facio-cutaneous syndrome: developmental consequences of germline Ras/MAPK activation on myogenesis. Am J Med Genet C Semin Med Genet. 2011;157C:104–14. [PubMed: 21495178]
- van der Burgt I, Kupsky W, Stassou S, Nadroo A, Barroso C, Diem A, Kratz CP, Dvorsky R, Ahmadian MR, Zenker M. Myopathy caused by HRAS germline mutations: implications for disturbed myogenic differentiation in the presence of constitutive HRas activation. J Med Genet. 2007;44:459–62. [PMC free article: PMC2598013] [PubMed: 17412879]
- van Steensel MA, Vreeburg M, Peels C, van Ravenswaaij-Arts CM, Bijlsma E, Schrander-Stumpel CT, van Geel M. Recurring HRAS mutation G12S in Dutch patients with Costello syndrome. Exp Dermatol. 2006;15:731-4. [PubMed: 16881968]
- Weaver KN, Care M, Wakefield E, Zarate YA, Skoch J, Gripp KW, Prada CE. Craniosynostosis is a feature of Costello syndrome. Am J Med Genet A. 2022;188:1280-6. [PubMed: 34964243]
- Weaver KN, Wang D, Cnota J, Gardner N, Stabley D, Sol-Church K, Gripp KW, Witte DP, Bove KE, Hopkin RJ. Early-lethal Costello syndrome due to rare HRAS tandem base substitution (c.35_36GC>AA; p.G12E) associated pulmonary vascular disease. Pediatr Dev Pathol. 2014;17:421–430. [PMC free article: PMC4294968] [PubMed: 25133308]
- White SM, Graham JM Jr, Kerr B, Gripp K, Weksberg R, Cytrynbaum C, Reeder JL, Stewart FJ, Edwards M, Wilson M, Bankier A. The adult phenotype in Costello syndrome. Am J Med Genet A. 2005;136:128–35. [PubMed: 15940703]
- Zampino G, Mastroiacovo P, Ricci R, Zollino M, Segni G, Martini-Neri ME, Neri G. Costello syndrome: further clinical delineation, natural history, genetic definition, and nosology. Am J Med Genet. 1993;47:176–83. [PubMed: 8213903]
Publication Details
Author Information and Affiliations
Nemours Children's Hospital
Wilmington, Delaware
T Jefferson University and Medical College
Philadelphia, Pennsylvania
Division of Human Genetics
Department of Pediatrics
Cincinnati Children's Hospital Medical Center and University of Cincinnati College of Medicine
Publication History
Initial Posting: August 29, 2006; Last Update: December 21, 2023.
Copyright
GeneReviews® chapters are owned by the University of Washington. Permission is hereby granted to reproduce, distribute, and translate copies of content materials for noncommercial research purposes only, provided that (i) credit for source (http://www.genereviews.org/) and copyright (© 1993-2024 University of Washington) are included with each copy; (ii) a link to the original material is provided whenever the material is published elsewhere on the Web; and (iii) reproducers, distributors, and/or translators comply with the GeneReviews® Copyright Notice and Usage Disclaimer. No further modifications are allowed. For clarity, excerpts of GeneReviews chapters for use in lab reports and clinic notes are a permitted use.
For more information, see the GeneReviews® Copyright Notice and Usage Disclaimer.
For questions regarding permissions or whether a specified use is allowed, contact: ude.wu@tssamda.
Publisher
University of Washington, Seattle, Seattle (WA)
NLM Citation
Gripp KW, Weaver KN. HRAS-Related Costello Syndrome. 2006 Aug 29 [Updated 2023 Dec 21]. In: Adam MP, Feldman J, Mirzaa GM, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2024.

