Summary
Clinical characteristics.
The phenotypic spectrum of GARS1-associated axonal neuropathy ranges from GARS1 infantile-onset SMA (GARS1-iSMA) to GARS1 adolescent- or early adult-onset hereditary motor/sensory neuropathy (GARS1-HMSN).
- GARS1-iSMA. Age of onset ranges from the neonatal period to the toddler years. Initial manifestations are typically respiratory distress, poor feeding, and muscle weakness (distal greater than proximal). Weakness is slowly progressive, ultimately requiring mechanical ventilation and feeding via gastrostomy tube.
- GARS1-HMSN. Age of onset is most commonly during the second decade (range eight to 36 years). Initial manifestations are typically muscle weakness in the hands sometimes with sensory deficits. Lower limb involvement (seen in ~50% of individuals) ranges from weakness and atrophy of the extensor digitorum brevis and weakness of toe dorsiflexors to classic peroneal muscular atrophy with foot drop and a high steppage gait.
Diagnosis/testing.
The diagnosis of GARS1-associated axonal neuropathy is established in a proband with suggestive clinical findings and a heterozygous pathogenic variant in GARS1 identified by molecular genetic testing.
Management.
Treatment of manifestations:
- GARS1-iSMA. Supportive treatment should be tailored to the needs of the affected individual and his/her current functional status (non-sitter, sitter, or walker). A multidisciplinary team to include a neurologist, pulmonologist, physiatrist, and medical geneticist is recommended.
- GARS1-HMSN. Symptomatic treatment includes facilitating activities of daily living and addressing of mobility needs by a multidisciplinary team that includes neurologists, physiatrists, orthopedic surgeons, and physical and occupational therapists.
Surveillance:
- GARS1-iSMA. Routine monitoring of: growth, nutritional status, safety of oral feeding vs gastric tube feeding, respiratory status, need for assistive devices for activities of daily living and mobility, developmental progress and educational needs, and family need for social work support.
- GARS1-HMSN. Routine monitoring of neurologic findings, physical therapy and occupational therapy needs, and skin for pressure ulcers or sores and skin breakdown (particularly the feet, hips, and other pressure points).
Agents/circumstances to avoid: Medications that are toxic or potentially toxic to persons with HMSN.
Evaluation of relatives at risk: It is appropriate to clarify the genetic status of apparently asymptomatic older and younger at-risk relatives of an individual with GARS1-HMSN in order to identify as early as possible those who would benefit from prompt initiation of symptomatic management and awareness of agents/circumstances to avoid.
Genetic counseling.
GARS1-associated axonal neuropathy is an autosomal dominant disorder.
- GARS1-iSMA. All probands reported to date with the GARS1-iSMA phenotype whose parents have undergone molecular genetic testing have the disorder as a result of a de novo GARS1 pathogenic variant. If the GARS1 pathogenic variant cannot be detected in the leukocyte DNA of either parent, the recurrence risk to sibs is estimated to be 1% because of the theoretic possibility of parental germline mosaicism.
- GARS1-HMSN. Most individuals diagnosed with the GARS1-HMSN phenotype have an affected parent. Each child of an individual with GARS1-HMSN is at a 50% risk of inheriting the GARS1 pathogenic variant.
Once the GARS1 pathogenic variant has been identified in an affected family member, prenatal testing for a pregnancy at increased risk and preimplantation genetic testing are possible.
GeneReview Scope
Table
GARS1 infantile-onset spinal muscular atrophy (GARS1-iSMA) GARS1 adolescent- or early adult-onset hereditary motor/sensory neuropathy (GARS1-HMSN) 2
Diagnosis
No consensus clinical diagnostic criteria for GARS1-associated axonal neuropathy have been published.
Suggestive Findings
GARS1-associated axonal neuropathy should be suspected in individuals with the following clinical findings; findings on EMG and neuroimaging; and family history.
Clinical Findings
GARS1 infantile-onset spinal muscular atrophy (GARS1-iSMA)
- Typically, infantile onset of respiratory distress, poor feeding, and muscle weakness, with distal weakness greater than proximal; however, some children may present with features similar to toddlers.
- Absence of molecular genetic findings of spinal muscular atrophy (SMA) (i.e., either biallelic SMN1 deletions or compound heterozygosity for an SMN1 deletion and an SMN1 sequence variant)
GARS1 adolescent- or early adult-onset hereditary motor/sensory neuropathy (GARS1- HMSN)
- Bilateral weakness and atrophy of thenar and first dorsal interosseous muscles with progression to involve hypothenar, foot, and peroneal muscles in many individuals and mild-to-moderate impairment of vibration sense developing in advanced illness in some individuals (see Figure 1)
- Sensory deficits including reduction of pinprick, temperature, touch, and vibration perception in a stocking and (less often) glove pattern

Electrophysiologic Studies
EMG shows denervation predominantly in the distal muscle groups at normal motor distal latencies and conduction velocities (see Table 1; pdf):
- Absent or markedly reduced (frequently <1 mV) compound muscle action potentials (CMAPs) are recorded from the abductor pollicis brevis (APB) by median nerve stimulation [Sivakumar et al 2005].
- Preserved CMAPs are recorded from the abductor digiti minimi (ADM) by ulnar nerve stimulation.
- CMAP amplitude recorded by stimulation of the peroneal nerve is <2 mV in most individuals and <1 mV in individuals having clinically evident leg atrophy.
- Normal median SNAP amplitudes and conduction velocities are seen in most individuals, even those with mildly prolonged distal motor latency.
- In individuals with advanced disease, needle EMG shows no voluntary motor activity in the abductor pollicis and first dorsal interossei because of marked atrophy. Spontaneous activity is often seen in these muscles.
- The elicited sural SNAPs are preserved but with a reduced amplitude, despite sensory axonal loss identified histopathologically on examination of a sensory nerve from an individual with the CMT2D subtype; similar but milder changes were seen in individuals with dSMA-V.
Note: EMG is more widely available than nerve biopsy, which can be used in a single individual in a family or in diagnostically difficult cases.
Neuroimaging. Magnetic resonance imaging of the spine may demonstrate volume loss of ventral nerve roots, best appreciated at the cauda equina. Brain MRI is normal [Markovitz et al 2020].
Family history. The affected individual represents a simplex case (i.e., a single occurrence in a family) or has a family history consistent with autosomal dominant inheritance (e.g., affected males and females in multiple generations).
Establishing the Diagnosis
The diagnosis of GARS1-associated axonal neuropathy is established in a proband with suggestive clinical findings and a heterozygous pathogenic variant in GARS1 identified by molecular genetic testing (see Table 2).
Note: Identification of a heterozygous GARS1 variant of uncertain significance does not itself establish or rule out the diagnosis of this disorder.
The molecular genetic testing approach will likely depend on the age of onset of disease manifestations.
For an infant or a toddler, initial testing is typically SMN1 molecular genetic testing for spinal muscular atrophy, followed by either a multigene panel or comprehensive genomic testing, which does not require the clinician to determine which gene is likely involved. Exome sequencing is most commonly used; genome sequencing is also possible.
For an introduction to comprehensive genomic testing click here. More detailed information for clinicians ordering genomic testing can be found here.
Note that commercial multigene panels for early-onset neuropathies may often not include GARS1.
For individuals presenting in adolescence or adulthood, a hereditary motor and sensory neuropathy (also called Charcot-Marie-Tooth [CMT] hereditary neuropathy or distal hereditary neuropathy) multigene panel that includes GARS1 and other genes of interest (see Differential Diagnosis) is most likely to identify the genetic cause of the condition while limiting identification of variants of uncertain significance and pathogenic variants in genes that do not explain the underlying phenotype. Note: (1) The genes included in the panel and the diagnostic sensitivity of the testing used for each gene vary by laboratory and are likely to change over time. (2) Some multigene panels may include genes not associated with the condition discussed in this GeneReview. (3) In some laboratories, panel options may include a custom laboratory-designed panel and/or custom phenotype-focused exome analysis that includes genes specified by the clinician. (4) Methods used in a panel may include sequence analysis, deletion/duplication analysis, and/or other non-sequencing-based tests.
For an introduction to multigene panels click here. More detailed information for clinicians ordering genetic tests can be found here.
Table 2.
Molecular Genetic Testing Used in GARS1-Associated Axonal Neuropathy
Clinical Characteristics
Clinical Description
Individuals with GARS1-associated axonal neuropathy can present across the life span (infancy through adulthood). Although in utero presentation of a fetus with a GARS pathogenic variant has not been reported to date, it is possible (if not likely) given reports of in utero presentations of spinal muscular atrophy [MacLeod et al 1999, Kong et al 2021]. GARS1 infantile-onset SMA presents with respiratory distress, poor feeding, and muscle weakness that is distal greater than proximal. GARS1 adolescent- or early adult-onset hereditary motor/sensory neuropathy presents from childhood to adulthood, most commonly with muscle weakness in the hands and sometimes with sensory deficits in a stocking and (less often) glove pattern.
GARS1 Infantile-Onset Spinal Muscular Atrophy (GARS1-iSMA)
Age of onset ranges from the neonatal period to the toddler years. The presenting manifestations are typically respiratory distress, poor feeding, and muscle weakness (distal weakness greater than proximal).
Neonates present emergently with respiratory distress (stridor, weak cry, and respiratory insufficiency), poor feeding, and severe hypotonia, ultimately requiring mechanical ventilation and early placement of a gastrostomy tube. In neonates, the neurologic examination is notable for hypotonia, hyporeflexia, and tongue fasciculations in some.
Infants typically have delayed motor milestones with subsequent motor milestone regression. They do not achieve independent walking [James et al 2006, Eskuri et al 2012, Liao et al 2015]. Toddlers present with delayed walking and lack of stability progressing to loss of ambulation [Liao et al 2015]. Infant and toddler examinations are notable for hypotonia, absent or diminished reflexes, and stridor in some.
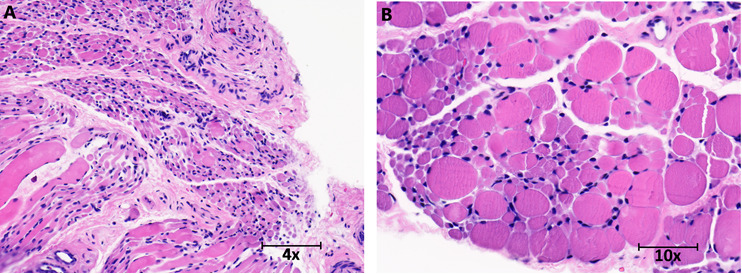
Nerve/muscle biopsy. Hematoxylin and eosin stain of quadriceps muscle of an infant show marked variation in fiber size with small group and fascicular atrophy typical of spinal muscular atrophy (see Figure 2).

GARS1 Adolescent- or Early Adult-Onset Hereditary Motor/Sensory Neuropathy (GARS1-HMSN)
Age of onset ranges from eight to 36 years, with most individuals (75%) developing manifestations during the second decade of life [Sivakumar et al 2005, James et al 2006].
The presenting manifestation is typically muscle weakness, often initially evident as transient cramping and pain in the hands on exposure to cold and cramping in calf muscles on exertion. Progressive weakness and atrophy of the thenar and first dorsal interosseus muscles are the major complaints (Figure 1). The hypothenar eminence is spared until later in the disease course.
The lower limbs are involved in about 50% of affected individuals. Lower-extremity involvement ranges from weakness and atrophy of the extensor digitorum brevis and weakness of toe dorsiflexors to classic peroneal muscular atrophy with foot drop and a high steppage gait. Peroneal muscles are affected earlier and more severely than the calf muscles. Peroneal muscular atrophy is associated with pes cavus and moderate sensory abnormalities. Reflexes at the ankles are diminished or absent in individuals with leg muscle weakness and sensory deficits.
Proximal limb muscle weakness is not observed in the upper or lower extremities.
Sensory examination is either normal or shows mild-to-moderate impairment of vibration sense in the hands and feet and reduction of pinprick, temperature, touch, and vibration perception in a stocking and (less often) glove pattern.
A minority of individuals show upper motor neuron signs (mild pyramidal signs and spasticity) [Christodoulou et al 1995, Sivakumar et al 2005, Dubourg et al 2006]. The occurrence of upper motor neuron signs may be attributed to relative preservation of sensory nerves and involvement of central motor pathways.
While progression of manifestations is often slow, extending over decades, some individuals may have a more rapid evolution of lower extremity involvement, which may more often be seen in the setting of both motor and sensory changes [Dubourg et al 2006].
Genotype-Phenotype Correlations
GARS1-iSMA. GARS1 pathogenic variants in the catalytic or anticodon binding domain are associated with this phenotype: p.Gly652Arg, p.Ile334Asn, p.Gly652Ala, and p.Asp200Tyr.
GARS1 adolescent- or early adult-onset HMSN
- GARS1 variants associated exclusively with distal spinal muscular atrophy V (dSMA-V): p.Leu183Pro and p.His472Arg.
- GARS1 variants associated with Charcot-Marie-Tooth neuropathy type 2D (CMT2D): p.Gly294Arg, p.Ile334Phe, and p.Gly580Arg.
Penetrance
To the authors' knowledge, reduced penetrance has not been described for GARS1-iSMA.
For adolescent- or adult-onset GARS1-HMSN, variable expressivity is described and at least one example of non-penetrance has been reported [Yalcouyé et al 2019].
Nomenclature
GARS1 infantile-onset spinal muscular atrophy (GARS1-iSMA). In this GeneReview, GARS1-iSMA is used to denote early-onset SMA (from the neonatal period to toddler age, presenting with respiratory insufficiency, poor feeding, hypotonia and areflexia).
GARS1 adolescent- or early adult-onset hereditary motor/sensory neuropathy (GARS1-HMSN)
- Charcot-Marie-Tooth neuropathy type 2D (CMT2D) and distal spinal muscular atrophy V (dSMA-V) were originally thought to be distinct entities. However, subsequent family studies [Sambuughin et al 1998] and later molecular genetic studies [Antonellis et al 2003] determined that they represent the clinical spectrum of adolescent- or early adult-onset HMSN caused by pathogenic variants in GARS1. CMT2D and dSMA-V are now collectively referred to as GARS1-HMSN. (See also CMT Hereditary Neuropathy Overview, Nomenclature.)
- CMT2D – characterized by distal motor and sensory neuropathy – may also be referred to as GARS1-associated distal motor and sensory neuropathy or, using the classification system proposed by Magy et al [2018], AD-CMTAx-GARS1.
- dSMA-V – characterized exclusively by distal motor involvement – may also be referred to as GARS1-associated distal hereditary motor neuropathy (GARS1-dHMN).
Prevalence
Disease prevalence is unknown; GARS1-associated axonal neuropathy is likely very rare. For example, fewer than 50 GARS1 pathogenic variants have been described; the vast majority are not recurrent [Meyer-Schuman & Antonellis 2017, Markovitz et al 2020].
Genetically Related (Allelic) Disorders
Biallelic germline pathogenic variants in GARS1 are associated with a multisystem phenotype that includes severe prenatal-onset growth restriction and developmental delays [Oprescu et al 2017].
Differential Diagnosis
Infantile-Onset Spinal Muscular Atrophy
Table 3.
Hereditary Disorders with Hypotonia in the Differential Diagnosis of GARS1 Infantile-Onset Spinal Muscular Atrophy (GARS1-iSMA)
Other inherited disorders to consider in the differential diagnosis of GARS1-iSMA include congenital myopathies (see X-Linked Centronuclear Myopathy) and metabolic/mitochondrial myopathies (see Glycogen Storage Diseases [GSD I, GSD II, GSD III, GSD IV, GSD V, GSD VI] and Mitochondrial Disorders Overview).
Infantile botulism may also resemble GARS1-iSMA. Like GARS1-iSMA, botulism in infants is associated with hyporeflexia, hypotonia, and muscle weakness. Unlike GARS1-iSMA, botulism is also associated with proximal weakness.
Adolescent- or Early Adult-Onset Hereditary Motor/Sensory Neuropathy (Charcot-Marie-Tooth Hereditary Neuropathy)
GARS1 adolescent- or early adult-onset hereditary motor/sensory neuropathy (HMSN) needs to be distinguished from other forms of HMSNs characterized by distal muscular atrophy, loss of reflexes, sensory deficits, reduced sensory nerve action potentials (SNAPs), and normal or mildly slowed motor nerve conduction velocity. The unique pattern of hand involvement before leg involvement and preserved SNAPs helps distinguish GARS1 adolescent- or early adult-onset HMSN from other axonal HMSNs. (See Charcot-Marie-Tooth Hereditary Neuropathy Overview).
Management
No clinical practice guidelines for GARS1-associated axonal neuropathy have been published.
Evaluations Following Initial Diagnosis
To establish the extent of disease and needs in an individual diagnosed with GARS1-associated axonal neuropathy, the evaluations summarized in Tables 4 and 5 (if not performed as part of the evaluation that led to the diagnosis) are recommended.
Table 4.
Recommended Evaluations Following Initial Diagnosis in Individuals with GARS1-Associated Axonal Neuropathy: Infantile-Onset Spinal Muscular Atrophy (GARS1-iSMA)
Table 5.
Recommended Evaluations Following Initial Diagnosis in Individuals with GARS1-Associated Axonal Neuropathy: Adolescent-/Adult-Onset Hereditary Motor/Sensory Neuropathy
Treatment of Manifestations
GARS1 Infantile-Onset Spinal Muscular Atrophy (GARS1-iSMA)
Supportive treatment of children with GARS1-iSMA should be individualized to the needs of the affected individual and his/her current functional status (non-sitter, sitter, or walker). A multidisciplinary team to include a neurologist, pulmonologist, physiatrist, and medical geneticist is recommended.
Table 6.
Supportive Treatment of Manifestations in Individuals with GARS1-Associated Axonal Neuropathy: Infantile-Onset Spinal Muscular Atrophy
GARS1 Adolescent- or Early Adult-Onset Hereditary Motor/Sensory Neuropathy (GARS1-HMSN)
Treatment is symptomatic. Affected individuals are often managed by a multidisciplinary team that includes neurologists, physiatrists, orthopedic surgeons, and physical and occupational therapists.
All individuals require assessment of their mobility needs and type of adaptations and devices that can be implemented to enhance their mobility. Devices include orthotics, which are often needed to correct foot drop and aid in walking.
Daily heel cord stretching helps prevent Achilles tendon shortening, foot deformities, and contractures.
Orthopedic surgery may be required for ankle fusion or to correct severe pes cavus deformity.
Some individuals require devices to assist with stability and mobility.
Numerous devices are available to facilitate various activities of daily living.
Surveillance
Table 7.
Recommended Surveillance for Individuals with GARS1-Associated Axonal Neuropathy: Infantile-Onset Spinal Muscular Atrophy
Table 8.
Recommended Surveillance for Individuals with GARS1-Associated Axonal Neuropathy: Adolescent-/Adult-Onset Hereditary Motor/Sensory Neuropathy
Agents/Circumstances to Avoid
Medications that are toxic or potentially toxic to persons with HMSN comprise a spectrum of risk ranging from definite high risk to negligible risk. See the Charcot-Marie-Tooth Association website (pdf) for an up-to-date list.
Chemotherapy for cancer that includes vincristine may be especially damaging to peripheral nerves and may severely worsen HMSN [Nishikawa et al 2008].
Given the relatively few individuals reported with GARS1-iSMA and limited longitudinal follow up, avoidance of neurotoxic (and potentially neurotoxic) medications would be appropriate despite the current lack of data.
Evaluation of Relatives at Risk
It is appropriate to clarify the genetic status of apparently asymptomatic older and younger at-risk relatives of an individual with GARS1-HMSN in order to identify as early as possible those who would benefit from prompt initiation of treatment and awareness of agents and circumstances to avoid.
See Genetic Counseling for issues related to testing of at-risk relatives for genetic counseling purposes.
Therapies Under Investigation
Search ClinicalTrials.gov in the US and EU Clinical Trials Register in Europe for access to information on clinical studies for a wide range of diseases and conditions. Note: There may not be clinical trials for this disorder.
Genetic Counseling
Genetic counseling is the process of providing individuals and families with information on the nature, mode(s) of inheritance, and implications of genetic disorders to help them make informed medical and personal decisions. The following section deals with genetic risk assessment and the use of family history and genetic testing to clarify genetic status for family members; it is not meant to address all personal, cultural, or ethical issues that may arise or to substitute for consultation with a genetics professional. —ED.
Mode of Inheritance
GARS1 infantile-onset spinal muscular atrophy is an autosomal dominant disorder; in all individuals reported to date, it has been caused by a de novo pathogenic variant.
GARS1 adolescent- or early adult-onset hereditary motor/sensory neuropathy (encompassing the phenotypes also referred to as Charcot-Marie-Tooth neuropathy type 2D and distal spinal muscular atrophy V) is an autosomal dominant disorder caused by an inherited or de novo pathogenic variant.
Risk to Family Members
GARS1 Infantile-Onset Spinal Muscular Atrophy (GARS1-iSMA)
Parents of a proband
- All probands reported to date with GARS1-iSMA whose parents have undergone molecular genetic testing have the disorder as a result of a de novo GARS1 pathogenic variant.
- Molecular genetic testing is recommended for the parents of the proband to confirm their genetic status and to allow reliable recurrence risk counseling.
- If the pathogenic variant identified in the proband is not identified in either parent, the following possibilities should be considered:
- The proband has a de novo pathogenic variant. Note: A pathogenic variant is reported as "de novo" if: (1) the pathogenic variant found in the proband is not detected in parental DNA; and (2) parental identity testing has confirmed biological maternity and paternity. If parental identity testing is not performed, the variant is reported as "assumed de novo" [Richards et al 2015].
- The proband inherited a pathogenic variant from a parent with germline (or somatic and germline) mosaicism. Note: Testing of parental leukocyte DNA may not detect all instances of somatic mosaicism and will not detect a pathogenic variant that is present in the germ cells only.
Sibs of a proband. The risk to sibs of the proband depends on the genetic status of the proband's parents:
- If a parent of the proband is known to have the GARS1 pathogenic variant identified in the proband, the risk to the sibs of inheriting the pathogenic variant is 50%.
- If the GARS1 pathogenic variant cannot be detected in the leukocyte DNA of either parent, the recurrence risk to sibs is estimated to be 1% because of the theoretic possibility of parental germline mosaicism [Rahbari et al 2016].
Offspring of a proband. Individuals with GARS1-iSMA are not known to reproduce; however, many are not yet of reproductive age.
Other family members. Given that all probands with GARS1-iSMA reported to date have the disorder as a result of a de novo GARS1 pathogenic variant, the risk to other family members is presumed to be low.
GARS1 Adolescent- or Early Adult-Onset Hereditary Motor/Sensory Neuropathy (GARS1-HMSN)
Parents of a proband
- Most individuals diagnosed with GARS1-HMSN have an affected parent.
- A proband with GARS1-HMSN may have the disorder as the result of a de novo pathogenic variant; the proportion of individuals with GARS1-HMSN caused by a de novo pathogenic variant is unknown.
- Molecular genetic testing is recommended for the parents of the proband to confirm their genetic status and to allow reliable recurrence risk counseling.
- If the pathogenic variant identified in the proband is not identified in either parent, the following possibilities should be considered:
- The proband has a de novo pathogenic variant. Note: A pathogenic variant is reported as "de novo" if: (1) the pathogenic variant found in the proband is not detected in parental DNA; and (2) parental identity testing has confirmed biological maternity and paternity. If parental identity testing is not performed, the variant is reported as "assumed de novo" [Richards et al 2015].
- The proband inherited a pathogenic variant from a parent with germline (or somatic and germline) mosaicism. Note: Testing of parental leukocyte DNA may not detect all instances of somatic mosaicism and will not detect a pathogenic variant that is present in the germ cells only.
- The family history of some individuals diagnosed with GARS1-HMSN may appear to be negative because of failure by health care professionals to recognize the syndrome, a milder phenotypic presentation, early death of a parent before the onset of symptoms, and/or late onset of the disorder. Therefore, an apparently negative family history cannot be confirmed unless molecular genetic testing has demonstrated that neither parent is heterozygous for the pathogenic variant identified in the proband.
Sibs of a proband. The risk to sibs of the proband depends on the clinical/genetic status of the proband's parents:
- If a parent of the proband is affected and/or is known to have the pathogenic variant identified in the proband, the risk to the sibs of inheriting the pathogenic variant is 50%.
- Intrafamilial clinical variability and reduced penetrance have been observed in GARS1-HMSN.
- If the GARS1 pathogenic variant cannot be detected in the leukocyte DNA of either parent, the recurrence risk to sibs is estimated to be 1% because of the theoretic possibility of parental germline mosaicism [Rahbari et al 2016].
- If the parents have not been tested for the GARS1 pathogenic variant but are clinically unaffected, the risk to the sibs of a proband appears to be low. However, sibs of a proband with clinically unaffected parents are still presumed to be at increased risk for GARS1-HMSN because of the possibility of reduced penetrance in a parent or the theoretic possibility of parental germline mosaicism.
Offspring of a proband. Each child of an individual with GARS1-HMSN is at a 50% risk of inheriting the GARS1 pathogenic variant.
Other family members. The risk to other family members depends on the genetic status of the proband's parents; if a parent is affected and/or has the GARS1 pathogenic variant, his or her family members are at risk.
Related Genetic Counseling Issues
Family planning
- The optimal time for determination of genetic risk and discussion of the availability of prenatal/preimplantation genetic testing is before pregnancy.
- It is appropriate to offer genetic counseling (including discussion of potential risks to offspring and reproductive options) to young adults who are affected or at risk.
Prenatal Testing and Preimplantation Genetic Testing
Once the GARS1 pathogenic variant has been identified in an affected family member, prenatal testing for a pregnancy at increased risk and preimplantation genetic testing are possible.
Differences in perspective may exist among medical professionals and within families regarding the use of prenatal testing. While most centers would consider use of prenatal testing to be a personal decision, discussion of these issues may be helpful.
Resources
GeneReviews staff has selected the following disease-specific and/or umbrella support organizations and/or registries for the benefit of individuals with this disorder and their families. GeneReviews is not responsible for the information provided by other organizations. For information on selection criteria, click here.
- Charcot-Marie-Tooth Association (CMTA)Phone: 800-606-2682Email: [email protected]
- European Charcot-Marie-Tooth ConsortiumDepartment of Molecular GeneticsUniversity of AntwerpAntwerp Antwerpen B-2610BelgiumFax: 03 2651002Email: [email protected]
- Hereditary Neuropathy FoundationPhone: 855-435-7268 (toll-free); 212-722-8396Fax: 917-591-2758Email: [email protected]
- Muscular Dystrophy Association (MDA) - USAPhone: 833-275-6321Email: [email protected]
- RDCRN Patient Contact Registry: Inherited Neuropathies Consortium
Molecular Genetics
Information in the Molecular Genetics and OMIM tables may differ from that elsewhere in the GeneReview: tables may contain more recent information. —ED.
Table A.
GARS-Associated Axonal Neuropathy: Genes and Databases
Table B.
OMIM Entries for GARS-Associated Axonal Neuropathy (View All in OMIM)
Molecular Pathogenesis
The molecular pathology that underlies the spectrum of neuromuscular and sensory phenotypes observed in GARS1-associated axonal neuropathy is unclear.
The protein GARS, which functions as a homodimer, is a member of the ubiquitously expressed aminoacyl-tRNA synthetase family whose key function is charging tRNA with glycine. GARS comprises an N-terminal appended WHEP-TRS domain, a catalytic core, and a C-terminal anticodon binding domain. Variants in GARS that encode each of these domains lead to axonal neuropathy [Antonellis et al 2003, Griffin et al 2014]. Functional studies in mice [Seburn et al 2014], yeast [Griffin et al 2014], zebrafish [Malissovas et al 2016], and in vitro [Griffin et al 2014] have confirmed the importance of the GARS catalytic, anti-codon binding, and dimerization domains in disease.
While absence of phenotypes in heterozygous null variants in mice provides evidence against haploinsufficiency [Seburn et al 2014], several variants resulting in impaired dimerization, anti-codon binding, or catalytic activity of GARS have been shown to contribute to the molecular and phenotypic overlap of the two HMSN subtypes, Charcot-Marie-Tooth neuropathy type 2D (CMT2D) and distal spinal muscular atrophy V (dSMA-V) [Griffin et al 2014, Malissovas et al 2016].
An overview of the gene-disease association can be found here.
Mechanism of disease causation. Not established
Table 9.
Notable GARS1 Pathogenic Variants
Chapter Notes
Author History
Anthony Antonellis, PhD; University of Michigan Medical School (2018-2021)
Rajarshi Ghosh, PhD (2021-present)
Lev G Goldfarb, MD; National Institute of Neurological Disorders and Stroke (2006-2021)
Timothy Lotze, MD (2021-present)
Rebecca Markovitz, MD, PhD (2021-present)
Lorraine Potocki, MD (2021-present)
Kumaraswamy Sivakumar, MD; Barrow Neurological Institute, Phoenix (2006-2021)
Revision History
- 22 July 2021 (bp) Comprehensive update posted live
- 29 November 2018 (ha) Comprehensive update posted live
- 25 August 2011 (me) Comprehensive update posted live
- 30 January 2007 (lgg) Revision: sequence analysis clinically available for mutations in GARS
- 8 November 2006 (me) Review posted live
- 24 February 2006 (lgg) Original submission
References
Literature Cited
- Abe A, Hayasaka K. The GARS gene is rarely mutated in Japanese patients with Charcot-Marie-Tooth neuropathy. J Hum Genet. 2009;54:310–2. [PubMed: 19329989]
- Antonellis A, Ellsworth RE, Sambuughin N, Puls I, Abel A, Lee-Lin SQ, Jordanova A, Kremensky I, Christodoulou K, Middleton LT, Sivakumar K, Ionasescu V, Funalot B, Vance JM, Goldfarb LG, Fischbeck KH, Green ED. Glycyl tRNA synthetase mutations in Charcot-Marie-Tooth disease type 2D and distal spinal muscular atrophy type V. Am J Hum Genet. 2003;72:1293–9. [PMC free article: PMC1180282] [PubMed: 12690580]
- Bönnemann CG, Wang CH, Quijano-Roy S, Deconinck N, Bertini E, Ferreiro A, Muntoni F, Sewry C, Béroud C, Mathews KD, Moore SA, Bellini J, Rutkowski A, North KN. Members of International Standard of Care Committee for Congenital Muscular Dystrophies. Diagnostic approach to the congenital muscular dystrophies. Neuromuscul Disord. 2014;24:289–311. [PMC free article: PMC5258110] [PubMed: 24581957]
- Christodoulou K, Kyriakides T, Hristova AH, Georgiou DM, Kalaydjieva L, Yshpekova B, Ivanova T, Weber JL, Middleton LT. Mapping of a distal form of spinal muscular atrophy with upper limb predominance to chromosome 7p. Hum Mol Genet. 1995;4:1629–32. [PubMed: 8541851]
- Cortese A, Wilcox JE, Polke JM, Poh R, Skorupinska M, Rossor AM, Laura M, Tomaselli PJ, Houlden H, Shy ME, Reilly MM. Targeted next-generation sequencing panels in the diagnosis of Charcot-Marie-Tooth disease. Neurology. 2020;94:e51–e61. [PMC free article: PMC7011687] [PubMed: 31827005]
- Del Bo R, Locatelli F, Corti S, Scarlato M, Ghezzi S, Prelle A, Fagiolari G, Moggio M, Carpo M, Bresolin N, Comi GP. Coexistence of CMT-2D and distal SMA-V phenotypes in an Italian family with a GARS gene mutation. Neurology. 2006;66:752–4. [PubMed: 16534118]
- Dubourg O, Azzedine H, Yaou RB, Pouget J, Barois A, Meininger V, Bouteiller D, Ruberg M, Brice A, LeGuern E. The G526R glycyl-tRNA synthetase gene mutation in distal hereditary motor neuropathy type V. Neurology. 2006;66:1721–6. [PubMed: 16769947]
- Eskuri JM, Stanley CM, Moore SA, Mathews KD. Infantile onset CMT2D/dSMA V in monozygotic twins due to a mutation in the anticodon-binding domain of GARS. J Peripher Nerv Syst. 2012;17:132–4. [PMC free article: PMC3572939] [PubMed: 22462675]
- Griffin LB, Sakaguchi R, McGuigan D, Gonzalez MA, Searby C, Züchner S, Hou YM, Antonellis A. Impaired function is a common feature of neuropathy-associated glycyl-tRNA synthetase mutations. Hum Mutat. 2014;35:1363–71. [PMC free article: PMC4213347] [PubMed: 25168514]
- Guenther UP, Varon R, Schlicke M, Dutrannoy V, Volk A, Hübner C, von Au K, Schuelke M. Clinical and mutational profile in spinal muscular atrophy with respiratory distress (SMARD): defining novel phenotypes through hierarchical cluster analysis. Hum Mutat. 2007;28:808–15. [PubMed: 17431882]
- He W, Bai G, Zhou H, Wei N, White NM, Lauer J, Liu H, Shi Y, Dumitru CD, Lettieri K, Shubayev V, Jordanova A, Guergueltcheva V, Griffin PR, Burgess RW, Pfaff SL, Yang XL. CMT2D neuropathy is linked to the neomorphic binding activity of glycyl-tRNA synthetase. Nature. 2015;526:710–4. [PMC free article: PMC4754353] [PubMed: 26503042]
- James PA, Cader MZ, Muntoni F, Childs AM, Crow YJ, Talbit K. Severe childhood SMA and axonal CMT due to anticodon binding domain mutations in the GARS gene. Neurology. 2006;67:1710–2. [PubMed: 17101916]
- Kong L, Valdivia DO, Simon CM, Hassinan CW, Delestrée N, Ramos DM, Park JH, Pilato CM, Xu X, Crowder M, Grzyb CC, King ZA, Petrillo M, Swoboda KJ, Davis C, Lutz CM, Stephan AH, Zhao X, Weetall M, Naryshkin NA, Crawford TO, Mentis GZ, Sumner CJ. Sci. Transl. Med. 2021;13:eabb6871. [PMC free article: PMC8208236] [PubMed: 33504650]
- Liao YC, Liu YT, Tsai PC, Chang CC, Huang YH, Soong BW, Lee YC. Two Novel De Novo GARS mutations cause early-onset axonal Charcot-Marie-Tooth disease. PLoS One. 2015;10:e0133423. [PMC free article: PMC4526224] [PubMed: 26244500]
- Lin S, Xu LQ, Xu GR, Guo LL, Lin BJ, Chen WJ, Wang N, Lin Y, He J. Whole exome sequencing reveals a broader variant spectrum of Charcot-Marie-Tooth disease type 2. Neurogenetics. 2020;21:79–86. [PubMed: 31832804]
- MacLeod MJ, Taylor JE, Lunt PW, Mathew CG, Robb SA. Prenatal onset spinal muscular atrophy. Eur J Paediatr Neurol. 1999;3:65–72. [PubMed: 10700541]
- Magy L, Mathis S, Le Masson G, Goizet C, Tazir M, Vallat JM. Updating the classification of inherited neuropathies: results of an international survey. Neurology. 2018;90:e870–e876. [PubMed: 29429969]
- Malissovas N, Griffin LB, Antonellis A, Beis D. Dimerization is required for GARS-mediated neurotoxicity in dominant CMT disease. Hum Mol Genet. 2016;25:1528–42. [PMC free article: PMC4805310] [PubMed: 27008886]
- Markovitz R, Ghosh R, Kuo ME, Hong W, Lim J, Bernes S, Manberg S, Crosby K, Tanpaiboon P, Bharucha-Goebel D, Bonnemann C, Mohila CA, Mizerik E, Woodbury S, Weimin B, Lotze T, Antonellis A, Xiao R, Potocki L. GARS-related disease in infantile spinal muscular atrophy: implications for diagnosis and treatment. Am J Med Genet Part A. 2020;182:1167–76. [PMC free article: PMC8297662] [PubMed: 32181591]
- Meyer-Schuman R, Antonellis A. Emerging mechanisms of aminoacyl-tRNA synthetase mutations in recessive and dominant human disease. Hum Mol Genet. 2017;26:R114–R127. [PMC free article: PMC5886470] [PubMed: 28633377]
- Nishikawa T, Kawakami K, Kumamoto T, Tonooka S, Abe A, Hayasaka K, Okamoto Y, Kawano Y. Severe neurotoxicities in a case of Charcot-Marie-Tooth disease type 2 caused by vincristine for acute lymphoblastic leukemia. J Pediatr Hematol Oncol. 2008;30:519–21. [PubMed: 18797198]
- Oprescu SN, Chepa-Lotrea X, Takase R, Golas G, Markello TC, Adams DR, Toro C, Gropman AL, Hou YM, Malicdan MCV, Gahl WA, Tifft CJ, Antonellis A. Compound heterozygosity for loss-of-function GARS variants results in a multisystem developmental syndrome that includes severe growth retardation. Hum Mutat. 2017;38:1412–20. [PMC free article: PMC5599332] [PubMed: 28675565]
- Rahbari R, Wuster A, Lindsay SJ, Hardwick RJ, Alexandrov LB, Turki SA, Dominiczak A, Morris A, Porteous D, Smith B, Stratton MR, Hurles ME, et al. Timing, rates and spectra of human germline mutation. Nat Genet. 2016;48:126–33. [PMC free article: PMC4731925] [PubMed: 26656846]
- Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, Grody WW, Hegde M, Lyon E, Spector E, Voelkerding K, Rehm HL., ACMG Laboratory Quality Assurance Committee. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17:405–24. [PMC free article: PMC4544753] [PubMed: 25741868]
- Sambuughin N, Sivakumar K, Selenge B, Lee HS, Friedlich D, Baasanjav D, Dalakas MC, Goldfarb LG. Autosomal dominant distal spinal muscular atrophy type V (dSMA-V) and Charcot-Marie-Tooth disease type 2D (CMT2D) segregate within a single large kindred and map to a refined region on chromosome 7p15. J Neurol Sci. 1998;161:23–8. [PubMed: 9879677]
- Seburn KL, Morelli KH, Jordanova A, Burgess RW. Lack of neuropathy-related phenotypes in hint1 knockout mice. J Neuropathol Exp Neurol. 2014;73:693–701. [PMC free article: PMC4098130] [PubMed: 24918641]
- Sivakumar K, Kyriakides T, Puls I, Nicholson GA, Funalot B, Antonellis A, Sambuughin N, Ellsworth K, Beggs JL, Zamba-Papanicolaou E, Ionasescu V, Dalakas MC, Green ED, Fischbeck KH, Goldfarb LG. Phenotypic spectrum of disorders associated with glycyl-tRNA synthetase mutations. Brain. 2005;128:2304–14. [PubMed: 16014653]
- Stenson PD, Mort M, Ball EV, Evans K, Hayden M, Heywood S, Hussain M, Phillips AD, Cooper DN. The Human Gene Mutation Database: towards a comprehensive repository of inherited mutation data for medical research, genetic diagnosis and next-generation sequencing studies. Hum Genet. 2017;136:665–77. [PMC free article: PMC5429360] [PubMed: 28349240]
- Yalcouyé A, Diallo SH, Coulibaly T, Cissé L, Diallo S, Samassékou O, Diarra S, Coulibaly D, Keita M, Guinto CO, Fischbeck K, Landouré G, H3African Consortium. A novel mutation in the GARS gene in a Malian family with Charcot-Marie-Tooth disease. Mol Genet Genomic Med. 2019;7,7 e00782. [PMC free article: PMC6625146] [PubMed: 31173493]
Publication Details
Author Information and Affiliations
Publication History
Initial Posting: November 8, 2006; Last Update: July 22, 2021.
Copyright
GeneReviews® chapters are owned by the University of Washington. Permission is hereby granted to reproduce, distribute, and translate copies of content materials for noncommercial research purposes only, provided that (i) credit for source (http://www.genereviews.org/) and copyright (© 1993-2024 University of Washington) are included with each copy; (ii) a link to the original material is provided whenever the material is published elsewhere on the Web; and (iii) reproducers, distributors, and/or translators comply with the GeneReviews® Copyright Notice and Usage Disclaimer. No further modifications are allowed. For clarity, excerpts of GeneReviews chapters for use in lab reports and clinic notes are a permitted use.
For more information, see the GeneReviews® Copyright Notice and Usage Disclaimer.
For questions regarding permissions or whether a specified use is allowed, contact: ude.wu@tssamda.
Publisher
University of Washington, Seattle, Seattle (WA)
NLM Citation
Markovitz R, Ghosh R, Lotze T, et al. GARS1-Associated Axonal Neuropathy. 2006 Nov 8 [Updated 2021 Jul 22]. In: Adam MP, Feldman J, Mirzaa GM, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2024.